DNA测序结果分析比对(实例)
DNA测序结果分析比对(实例)
关键词:dna测序结果2013-08-22 11:59来源:互联网点击次数:14423
从测序公司得到的一份DNA测序结果通常包含.seq格式的测序结果序列文本和.ab1格式的测序图两个文件,下面是一份测序结果的实例:
CYP3A4-E1-1-1(E1B).ab1
CYP3A4-E1-1-1(E1B).seq
.seq文件可以用系统自带的记事本程序打开,.ab1文件需要用专门的软件打开。软件名称:Chromas
软件Chromas下载
.seq文件打开后如下图:
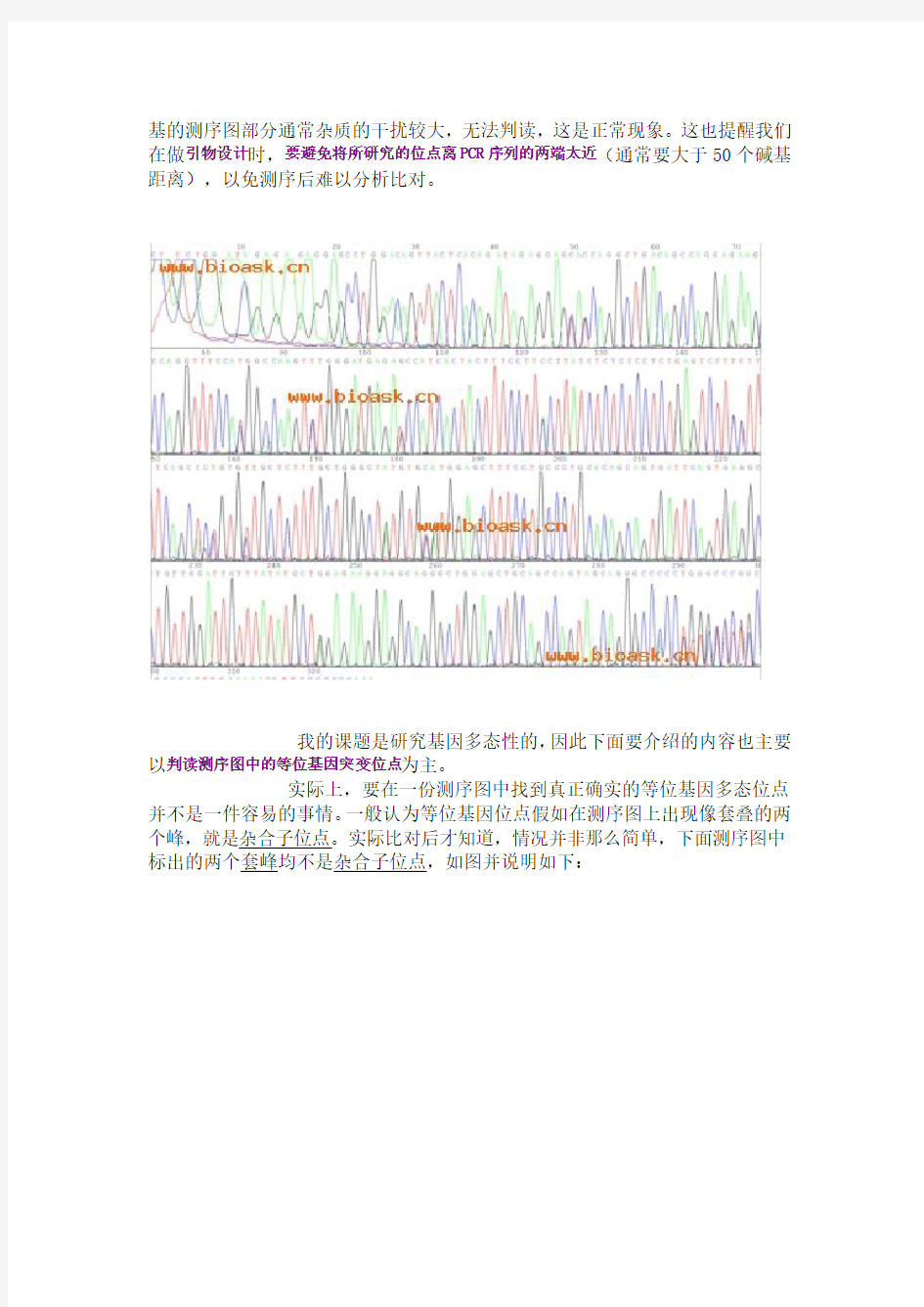
.ab1文件打开后如下图:
通常一份测序结果图由红、黑、绿和蓝色测序峰组成,代表不同的碱基序列。测序图的两端(下图原图的后半段被剪切掉了)大约50个碱
基的测序图部分通常杂质的干扰较大,无法判读,这是正常现象。这也提醒我们在做引物设计时,要避免将所研究的位点离PCR序列的两端太近(通常要大于50个碱基距离),以免测序后难以分析比对。
我的课题是研究基因多态性的,因此下面要介绍的内容也主要以判读测序图中的等位基因突变位点为主。
实际上,要在一份测序图中找到真正确实的等位基因多态位点并不是一件容易的事情。一般认为等位基因位点假如在测序图上出现像套叠的两个峰,就是杂合子位点。实际比对后才知道,情况并非那么简单,下面测序图中标出的两个套峰均不是杂合子位点,如图并说明如下:
说明:
第一组套峰,两峰的轴线并不在同一位置,左侧的T峰是干扰峰;第二组套峰,虽两峰轴线位置相同,但两峰的位置太靠近了,不是杂合子峰,蓝色的C峰是干扰峰通常的杂合子峰由一高一略低的两个轴线相同的峰组成,此处的序列被机器误判为“C”,实际的序列应为“A”,通常一个高大碱基峰的前面
1~2个位点很容易产生一个相同碱基的干扰峰,峰的高度大约是高大碱基峰的1/2,离得越近受干扰越大。
一个摸索出来的规律是:主峰通常在干扰峰的右侧,干扰峰并不一定比主峰低。最关键的一点是一定要拿疑似为杂合子峰的测序图位点与测序结果的文本序列和基因库中的比对结果相比较;一个位点的多个样本相比较;你得出的该位点的突变率与权威文献或数据库中的突变率相比较。
通常,对于一个疑似突变位点来说,即使是国际上权威组织大样本的测序结果中都没有报道的话,那么单纯通过测序结果就判定它是突变点,是并不严谨的,因一份 PCR产物中各个碱基的实际含量并不相同,很难避免不产生误差的。对于一个未知突变位点的发现,通常还需要用到更精确的酶切技术。
(责任编辑:大汉昆仑王)
DNA测序常见问题及分析
DNA测序过程可能遇到的问题及分析 对于一些生物测序公司(如Invitrogen等),我们的菌液或质粒经过PCR和酶切鉴定都没问题,但几天后的测序结果却无法另人满意。 为什么呢? PCR产物直接进行测序,在PCR产物长度以后将无反应信号,机器将产生许多N值。这是由于Taq酶能够在PCR反应的末端非特异性地加上一个A碱基,我们所用的T载体克隆PCR产物就是应用该原理,通常PCR产物结束的位点,PCR产物测序一般末端的一个碱基为A(绿峰),也就是双脱氧核甘酸ddNTP终止反应的位置之前的A,A后的信号会迅速减弱。 N值情况一般是由于有未去除的染料单体造成的干扰峰。该干扰峰和正常序列峰重叠在一起,有时机器377以下的测序仪无法正确判断出为何碱基。有时,在序列的起始端的小片段容易丢失,导致起始区信号过低,机器有时也无法正确判读。在序列的3’端易产生N值。一个测序反应一般可以读出900bp以上的碱基(ABI3730可以达到1200bp),但是,只有一般600bp以前的碱基是可靠的,理想条件下,多至700bp的碱基都是可以用的。一般在650bp以后的序列,由于测序毛细管胶的分辩率问题,会有许多碱基分不开,就会产生N值。测序模板本身含杂合序列,该情况主要发生在PCR产物直接测序,由于PCR产物本身有突变或含等位基因,会造成在某些位置上有重叠峰,产生N值。这种情况很容易判断,那就是整个序列信号都非常好,只有在个别位置有明显的重叠峰,视杂合度不同N值也不同。 测序列是从引物3’末端后第一个碱基开始的,所以就看不到引物序列。有两种方法可以得到引物序列。1.对于较短的PCR产物 (<600bp),可以用另一端的引物进行测序,从另一端测序可以一直测通,可以在序列的末端得到该引物的反向互补序列。对于较长的序列,一个测序反应测不通,就只能将PCR产物片段克隆到载体中,用载体上的通用引物(T7/SP6)进行测序。载体上的通用引物与所插入序列间
2_重测序BSA分析项目结题报告
重测序BSA项目结题报告 客户单位:____________________________________ 报告单位:____________ 联系人:____________________________________ 联系电话: ___________________________ 传真:___________________________ 报告日期:____________________________________ 项目负责人:__________ 审核人: __________________ 目录 目录 (1) 1 项目概况 (1) 1.1 合同关键指标 (1)
1.2 项目基本信息 (1) 1.3 项目执行情况 (2) 1.4项目结果概述 (2) 2 项目流程 (3) 2.1 实验流程 (3) 2.2 信息分析流程 (3) 3 生物信息学分析 (5) 3.1 测序数据质控 (5) 3.1.1 原始数据介绍 (5) 3.1.2 碱基测序质量分布 (7) 3.1.3碱基类型分布 (9) 3.1.4 低质量数据过滤 (10) 3.1.5测序数据统计 (10) 3.2 与参考基因组比对统计 (11) 3.2.1 比对结果统计 (11) 3.2.2 插入片段分布统计 (11) 3.2.3 深度分布统计 (12) 3.3 SNP 检测与注释 (14) 331样品与参考基因组间SNP的检测 (14) 332样品之间SNP的检测 (17) 3.3.3 SNP结果注释 (19) 3.4 Small In Del 检测与注释 (22) 3.4.1 样品与参考基因组间Small InDel 的检测 (22) 3.4.2样品之间Small InDel 检测 (22) 343 Small In Del 的注释 (23) 3.5 关联分析 (26) 3.5.1高质量SNP筛选 (26) 3.5.2 SNP-index方法关联结果 (26) 3.5.3 ED方法关联结果 (28)
人类基因组重测序分析
6 首页 科技服务 医学检测 科学与技术 市场与支持 加入我们 关于我们提供领先的基因组学解决方案 Providing Advanced Genomic Solutions 诺禾致源 人类疾病基因组重测序分析图3 Circos 图 人类基因组重测序分析6项升级 Novo-Zhonghua Genomes 数据库注释 一些位点的突变可能在千人基因组中或在欧美人群中属于低频突变,但是对于中国人群来说却是常见突变。诺禾致源自建中国人数据库 Novo-Zhonghua Genomes,数据库中的所有样本均来自正常中国人群。已有研究表明,与国际通用的多人种数据库相比,使用单一人种数据库进行疾病研究,可以有效减少假阳性现象。 图2 真核生物基因的结构[6] 复杂疾病变异分类标准 DamLevel Variant Calling Variant Annotation Benign Likely Benign VUS Likely Pathogenic Custom knowledge Clinical Data Pathogenic Family Testing Published + in house data Population frequency Predictions: PolyPhen, SIFT, etc Amino acid conservation Published Disease Information Variant classification Candidate Variants Novo-Zhonghua Genomes 数据库注释 复杂疾病突变位点有害性分类 非编码区(Non-coding region)分析 疾病基因组 CNV/SV 分析 基于基因(Gene-based)的 Burden Analysis (复杂疾病散发样本) 可视化的数据结果展示 基于健康中国人群的千人测序数据,测序深度 > 30× 参考 ACMG 等,推出针对复杂疾病变异位点有害性的分类标准 应用 ENCODE 数据库最新内容,并结合国际通用数据库、自建数 复杂疾病突变位点有害性分类 基于美国医学遗传学会 ACMG[2]与 Duzkale H[3]提出的变异分类标准,诺禾致源疾病基因组信息分析团队推出了一套针对复杂疾病变异位点有害性的分类标准 DamLevel(如下图所示)。DamLevel 将变异位点的有害性分为5个层级:Pathogenic、Likely Pathogenic、VUS(Variant of uncertain significance)、Likely Begnin、Begnin,更好地鉴定个体遗传变异与疾病的相关性。 非编码区(Non-coding region)分析 基因组非编码区变异可以引发多种疾病,包括心脏类疾病、糖尿病、癌症、肥胖症等[4,5],但目前对非编码区突变的筛选和功能描述仍具挑战性。诺禾致源非编码区分析,应用 ENCODE 数据库最新内容对非编码区突变进行注释,通过国际通用数据库和自建的 Novo-Zhonghua Genomes 数据库进行频率筛选以及保守性过滤,精确定位非编码区中低频且保守的突变,筛选到与疾病相关的非编码区突变。 疾病基因组 CNV/SV 分析 CNV/SV 与基因表达、表型、人类疾病发生发展都有着非常密切的关系[7,8],诺禾致源疾病基因组信息分析团队研发了一整套 CNV/SV 筛选方法,包括有害性 CNV/SV 筛选和 de novo CNV/SV 分析(基于成三或成四家系)等。利用 DGV、DECIPHER、CNVD 等数据库对变异检出结果进行标记,从结果中进一步过滤掉良性 CNV/SV,经过一系列筛选后,准确鉴定个体 CNV/SV 遗传变异与疾病的相关性。 图4 CNV 分布图 表1 本次产品升级亮点 图5 Burden 分析结果的热图展示 1 2 3 4 5 Novo-Zhonghua Genomes 数据库注释 Novo-Zhonghua Genomes 数据库是诺禾致源自建针对 中国正常人群的数据库,助 力中国人群基因组信息解析。 复杂疾病突变位点 有害性分类 诺禾致源推出的复杂疾病变 异位点有害性的分类标准 (DamLevel),准确标识复杂 疾病的致病性突变位点。 非编码区 (Non-coding region)分析 应用 ENCODE 数据库最新内 容对非编码区进行注释、筛 选,精确定位非编码区中低 频且保守的突变。 疾病基因组 CNV/SV 分析 完整的有害性 CNV/SV 筛选 和 de novo CNV/SV 分析, 准确鉴定个体 CNV/SV 遗传 变异与疾病的相关性。 基于基因 (Gene-based)的 Burden Analysis 针对复杂疾病的研究,通过 检测疾病状态与基因变异的 相关性,寻找特定疾病(或 性状)的易感基因。 可视化的 数据结果展示 灵活易用的测序数据结果展 示,使大量复杂数据的分析 变得轻松而高效,提高数据 可读性。 ? log 10 ( P ? value ) Mutations of Genes Prioritized by Burden Analysis CIR1 PIGP CTSE PRB2 CYP HDAC1 GRK6 PIGK MYL6B EHD2 0810 246 Mutations 4 3 2 1 基于基因(Gene-based)的 Burden Analysis 关联分析是研究复杂疾病的1个重要方法,其通过检测疾病状态与基因变异的相关性,寻找特定疾病(或性状)的易感基因。通常是在具有不同表型的2组个体(一般为患病者和正常对照者)中,基于遗传位点(或基因、单体型)的频率分布差异,间接反映该遗传位点(或基因)可能与疾病(或性状)存在关联性。 Burden Analysis(Gene-based)基于复杂疾病的 case 和 control 散发样本,通过 Fisher's exact test 以及 SKAT 统计方法分析得到候选基因,针对候选基因可以进行富集分析(KEGG 富集分析和 GO 富集分析)与蛋白网络互作分析。 可视化的结果展示 诺禾致源疾病基因组信息分析团队,会为客户提供不断更新的变异注释、项目特异性分析和灵活易用的“变异-基因-疾病”可视化结果,让科学研究更轻松。 图6 疾病与基因关联性展示图 产品名称升级亮点 引领行 业新 标杆 参考文献 [1] Nagasaki M, Yasuda J, Katsuoka F, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals.[J]. Nature Communications, 2015, 6. 阅读原文 >> [2] Richards S, Aziz N, Bale S, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genetics in Medicine, 2015. 阅读原文 >> [3] Duzkale H, Shen J, McLaughlin H, et al. A systematic approach to assessing the clinical significance of genetic variants[J]. Clinical genetics, 2013, 84(5): 453-463. 阅读原文 >> [4] Yoshinari M, Akihiko M, Dongquan S, et al. A functional polymorphism in the 5' UTR of GDF5 is associated with susceptibility to osteoarthritis.[J]. Nature Genetics, 2007, 39(4):529-33. 阅读原文 >> [5] Kjong-Van L, Ting C. Exploring functional variant discovery in non-coding regions with SInBaD.[J]. Nucleic Acids Research, 2012, 41 (1):e7-e7. 阅读原文 >> [6] https://https://www.360docs.net/doc/301620203.html,/wiki/Regulatory_sequence 阅读原文 >> [7] Sudmant P H, Rausch T, Gardner E J, et al. An integrated map of structural variation in 2,504 human genomes.[J]. Nature, 2015, 526 (7571):75-81. 阅读原文 >> [8] Birney E, Soranzo N. Human genomics: The end of the start for population sequencing.[J]. Nature, 2015, 526(7571):52-3. 阅读原文 >> 免费升级7-9月 新签合同 免费升级数据分析
一代测序常见问题及解决策略
测序常见问题及解决策略 一、PCR常见问题 1.假阴性,不出现扩增条带 PCR出现假阴性结果,可从以下几个方面来寻找原因: 1)模板:①模板中有杂蛋白;②模板中有Taq酶抑制剂;③在提取制备模板时丢失过多;④模板核酸变性不彻底。 2)酶:酶失活或反应时忘了加酶。 3)Mg2+浓度:Mg2+浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR 扩增产量甚至使PCR扩增失败而不出扩增条带。 4)反应条件:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。 5)靶序列变异:靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。 2.假阳性 假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。常见原因有: 1)引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。靶序列太短或引 物太短,容易出现假阳性。需重新设计引物。 2)靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。二是空气中的 小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。 3.出现非特异性扩增带 PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。非特异性条带的出现,其原因:一是引物
高通量测序NGS数据分析中的质控
高通量测序错误总结 一、生信分析部分 1)Q20/Q30 碱基质量分数与错误率是衡量测序质量的重要指标,质量值越高代表碱基被测错的概率越小。Q30代表碱基的正确判别率是99.9%,错误率为0.1%。同时我们也可以理解为1000个碱基里有1个碱基是错误的。Q20代表该位点碱基的正确判别率是99%,错误率为1%。对于整个数据来说,我们可以认为100个碱基里可能有一个是错误的, 在碱基质量模块报告的坐标图中,背景颜色沿y-轴将坐标图分为3个区:最上面的绿色是碱基质量很好的区,Q值在30以上。中间的橘色是碱基质量在一些分析中可以接受的区,Q值在20-30之间。最下面红色的是碱基质量很差的区。在一些生信分析中,比如以检查差异表达为目的的RNA-seq分析,一般要求碱基质量在Q在Q20以上就可以了。但以检查变异为目的的数据分析中,一般要求碱基质量要在Q30以上。 一般来说,测序质量分数的分布有两个特点: 1.测序质量分数会随着测序循环的进行而降低。 2.有时每条序列前几个碱基的位置测序错误率较高,质量值相对较低。 在图中这个例子里,左边的数据碱基质量很好,而右边的数据碱基质量就比较差,需要做剪切(trimming),根据生信分析的目的不同,要将质量低于Q20或者低于Q30的碱基剪切掉。 2)序列的平均质量 这个是碱基序列平均质量报告图。横坐标为序列平均碱基质量值,纵坐标代表序列数量。通过序列的平均质量报告,我们可以查看是否存在整条序列所有的碱基质量都普遍过低的情况。一般来说,当绝大部分碱基序列的平均质量值的峰值大于30,可以判断序列质量较好。如这里左边的图,我们可以判断样品里没有显着数量的低质量序列。但如果曲线如右边的图所示,在质量较低的坐标位置出现另外一个或者多个峰,说明测序数据中有一部分序列质量较差,需要过滤掉。 3)GC含量分布 这个是GC含量分布报告图。GC含量分布检查是检测每一条序列的GC含量。将样品序列的GC 含量和理论的GC含量分布图进行比较,用来检测样品数据是否有污染等问题。理论上,GC含量大致是正态分布,正态分布曲线的峰值对应基因组的GC含量。如果样品的GC含量分布图不是正态分布,如右图出现两个或者多个峰值,表明测序数据里可能有其他来源的DNA序列污染,或者有接头序列的二聚体污染。这种情况下,需要进一步确认这些污染序列的来源,然后将污染清除。 4)序列碱基含量
测序结果处理方法及聚类分析(DOC)
一、测得序列的拼接及处理 1、送样类型 a非克隆法(如PCR产物、PCR产物纯化回收等) 由于此类型样品,两端的引物序列一般在测序的过程中会有缺失,很难找全引物序列,仅能找到部分引物序列,因此对于此类型样品的测序结果可以不做引物序列的查找,后续需要可再做引物序列的查找。 b克隆法(片段通过TA克隆或其他载体构建等) 此类型样品,目的片段两端的引物可以很完整的保存在载体中,引物序列亦是测序片段,所以引物序列比较完整,可以找到引物的完整序列,因此可以通过查找引物序列而找到目的片段的起始位置。 2、测序方法 观察峰值图可用软件“bioedit” a单向测通 对于此种测序结果基本上单条序列不需要拼接,通过观察序列峰值图来初步判断序列结果的准确性,一般来说峰越尖越好,套峰越少越好。 b双向测通 对于此种测序结果,除了要观察峰值图的好坏外,要得到完整的序列,还需要对双向序列进行拼接,利用DNASTAR中seqMan进行拼接,点击“NEW”、“add sequence”(一般为abi格式,选择双向测序结果)、“assemble”,“contig”,一般保存完整的片段长度即选择“All”,亦可保存其中的片段长度,保存格式一般选择“fas”格式以便在不同的编辑软件中使用。具体步骤如下图。
3、对测得的序列进行比对及聚类分析 一般来讲,可以将所有需要进行比对的序列粘贴在一个记事本中,保存的格式最好 为“fas”格式,,利用软件“MEGA”中“Align”打开所需序列,依据序列的特性进行选择如DNA或protein,然后添加所有需要进行比对的序列。
可根据序列的具体情况进行选择比对的方法,本教程选择“ClustalW”法。 析,可保存为该软件格式,或其他格式。
20个测序常见的问题
20个测序常见的问题 1.为什么需要新鲜的菌液? 首先,新鲜的菌液易于培养,可以获得更多的DNA,同时最大限度地保证菌种的纯度。2.如何提供菌液? 如果您提供新鲜菌液,用封口膜封口以免泄漏;也可以将培养好的4~5ml菌液沉淀下来,倒去上清以方便邮寄。同时邮寄时最好用盒子以免邮寄过程中压破。 3.如何制作穿刺菌? 用灭菌过1.5ml或2ml离心管加入LB琼脂(7g/L)斜面凝固,用接种针挑取分散良好的单菌落穿过琼脂直达管底,不完全盖紧管盖适当温度培养过夜,然后盖紧盖子加封口膜,室温或4度保存。 4.PCR产物直接测序有什么要求? (1)扩增产物必须特异性扩增,条带单一。如果扩增产物中存在非特异性扩增产物,一般难以得到好的测序结果; (2)必须进行胶回收纯化; (3)DNA纯度在1.6—2.0之间,浓度50ng/ul以上。 5.为什么PCR产物直接测序必须进行Agarose胶纯化? 如果不进行胶纯化而直接用试剂盒回收,经常会导致测序出现双峰甚至乱峰,这主要是非特异性扩增产物或者原来的PCR引物去除不干净所导致。大多所谓的PCR“纯化试剂盒”实际上只是回收产物而不能起到纯化的作用的。对于非特异性扩增产物肯定无法去除,而且通常他们不能够完全去除所有的PCR引物,这会造成残留的引物在测序反应过程中参与反应而导致乱峰。 6.如何进行PCR产物纯化? PCR产物首先必须用Agarose胶电泳,将特异扩增的条带切割下,然后纯化。使用凝胶回收试剂盒回收,产物用ddH2O溶解。 7.PCR产物直接测序的好处? (1) PCR产物直接测序可以反映模板的真实情况; (2) 省去克隆的实验费用和时间; (3) PCR产物测序正确的片段进行下一步克隆实验使结果更有保障; (4) 混合模板进行PCR的产物直接测序可以发现其中的点突变。 8.对用于测序的质粒DNA的要求有哪些? 对测序模板DNA的一般要求:(1)DNA纯度要求高,1.6—2.0之间,不能有混合模板,也不能含有RNA,染色体DNA,蛋白质等;(2)溶于ddH2O中,溶液不能含杂质,如盐类,或EDTA等螯合剂,将干扰测序反应正常进行。 9.如何鉴定质粒DNA浓度和纯度? 我们使用水平琼脂糖凝胶电泳,并在胶中加入0.5ug/ml的EB(电泳缓冲液中不必加E,加一个已知浓度的标准样品。电泳结束以后在紫外灯下比较亮度,判断浓度和纯度。此方法可以更直接、准确地判断样品中是否含有染色体DNA、RNA等,也可以鉴别抽提的质粒DNA 的不同构型。 质粒DNA的3种构型是指在抽提质粒DNA过程中,由于各种原因的影响,使得超螺旋的共价闭合环状结构的质粒(SC)的一条链断裂,变成开环状(OC)分子,如果两条链发生断裂,就变成为线状(L)分子。这3种分子有不同的迁移率,通常,超螺旋型(SC)迁移速度最快,其次为线状(L)分子,最慢为开环状(OC)分子。使用紫外分光光度计检测,或者用溴乙锭-标准浓度DNA比较法只能检测抽提到的产物中的浓度,甚至由于抽提的质粒DNA中含有RNA、蛋白质、染色体DNA等因素的干扰,浓度检测的数值也是没有多少意义的。
序列拼接
序列拼接 * 为了保证测序结果的准确性,单基因短片段(700pd左右)测序一般应采用双向测序,然后将双向测序的结果拼接在一起,从而获得一致性序列。线粒体基因组测序和DNA长片段测序一般是通过分段测序来完成的,最后也需要将测出的短片段拼接成一条完整的序列。序列拼接可以在不同的软件中进行。 一、使用“组装批处理文件byLHM.pg4”进行拼接 1. 在预定的位置建立一个文件夹“gap”,将需要使用的3个软件“组装批处理文件byLHM.pg4”、“V ector_primer4pMD18-T.vec_pri”、“pMD18-T_Vector.seq”拷贝到该文件夹下,再将需要拼接的测序文件拷贝到该文件夹下。 2. 双击运行“组装批处理文件byLHM.pg4”程序。 3. 在程序运行后出现的界面右侧点击“Add files”按钮,打开要拼接的序列文件。为了保证 拼接后输出的是正向序列,最好先添加上游引物序列,然后添加下游引物序列,因为在一般情况下软件将添加的第一条序列默认为正向参照序列;有时由于测序效果等因素的影响,有时即使首先添加的是上游引物序列,但拼接后仍然会以测序效果明显更好的下游引物序列为正向参照序列,此时需要按照后面介绍的方法将上游引物序列转换为正向参照序列再输出一致性序列。 4. 点击界面上方第二行的“Configure Modules”,在弹出的窗口左边的任务栏中点击“[x] Sequencing vector Clip”,再点击右边的“Browse”按钮,通过弹出的窗口打开“Vector_primer4pMD18-T.vec_pri”程序;点击左边任务栏中的“[] Cloning Vector Clip”,再点击右边的“Browse”按钮,通过弹出的窗口打开“pMD18-T_Vector.seq”程序;点击左下角的“Run”按钮,即开始数据处理,处理结果将自动保存到“gap”文件夹中。 5. 在“gap”文件夹中双击“AssMit_tmp.o.aux”文件,将鼠标移到弹出的“Contig Selector” 窗口中的直线上,点击右键,选择“Edit Contig”,即弹出“Contig Editor”窗口,点击最右边的“setting”按钮,在下拉菜单中选择“By background colour”,即可显示比对结果的有差异碱基;双击某一序列,即可显示该序列的测序峰图,以检查核对该位点碱基的测序情况。 * 注:执行此操作时一定要检查正向序列是否为上游引物序列;如果不是,则需要将上游引物序列转换成正向序列后再执行下面的“输出及保存序列”操作;具体的操作步骤是:点击“GAPv4.10 AssMit_tmp.o”窗口中的“Edit”菜单,在下拉菜单中选择“Complement a contig”命令,在弹出来的“Complement contig”小窗口中检查确认“Contig identifier” 框中的序列为上游引物序列,然后点击“OK”即将完成序列转换。 6. 点击“GAPv4.10 AssMit_tmp.o”窗口中的“File”菜单,在下拉菜单中选择“Save consensus”可保存一致序列,nomors------ok ,序列即保存在刚刚使用过的那个文件夹中,然后把文件名改成用“*.txt”形式,以便保存的文件成为文本文件,若忘记在文件名后加“.txt”,则保存完毕后可将文件的扩展名改成“.txt”;只有拼接好的一致序列才可用于后面的序列分析。 7.然后把在ncbi里查到的相近种的序列放到一起,也可以直接放到刚才那个cons.txt文本文 档中,然后打开clustalx.exe进行序列比对,file------load sequence ------G盘-----004文件夹-----cons.txt-----aligenment-----do complete aligenment,这时如果发现两条序列的保守区域很不对,极可能是刚刚测得这个种的序列反了,需要用Bioedit把它正过来, 8.在程序里打开已经安装好的Bioedit,例如找file---------open----G盘---004----cons.txt,打开, 选sequence--------下拉菜单中找Nuclic acid,在菜单中找reverse complement,点击它 然后在另一对话框中例如G:/004/CONS.TXT中点击保存save Aligenment. 这样序列即
CHIP SEQ分析常见问题集锦
ChIP-Seq分析常见问题集锦 染色质免疫共沉淀测序(ChIP-Seq)是指对染色质免疫共沉淀(ChIP)获得的DNA片段进行大规模测序,并能把所研究蛋白的DNA结合位点精确定位到基因组上。 Roche GS FLX Titanium、Illumina Solexa GA IIx和AB SOLID4这3种测序技术均可以用于ChIP-seq,其中采用Illumina Solexa GA IIx进行ChIP-Seq已有较多文献报道。 ChIP-Seq技术高质量、高通量、低成本的数据产出,为表观遗传组学研究奠定了技术基础。研究者可以在以下几方面展开研究:(1)判断DNA链的某一特定位置会出现何种组蛋白修饰;(2)检测RNA polymerase II及其它反式因子在基因组上结合位点的精确定位;(3)研究组蛋白共价修饰与基因表达的关系;(4)CTCF转录因子研究。 ChIP-Seq有什么样品要求? 答:(1)请提供浓度≥10ng/ul、总量≥200ng、OD260/280为1.8~2.2的DNA样品;若单次ChIP后DNA量不够,建议将2~3次ChIP的DNA合并在一起。 (2)请提供DNA打断时检测胶图,要求打断后DNA电泳主带在200-500bp范围内;请对于ChIP 获得DNA设计引物进行QPCR验证和定量,能够提供检测位点的检测报告。附阳性和阴性对照。(3)样品请置于1.5ml管中,管上注明样品名称、浓度以及制备时间,管口使用Parafilm 封口。在运输前将所有样品管固定于50ml带盖离心管中,再将50ml管放在封口袋中。 ChIP-Seq相比ChIP-chip有哪些优势? 答:第一,ChIP-Seq能实现真正的全基因组分析。目前所能获得的芯片上固定的探针只能代表全基因组部分序列,所获得的杂交信息具有偏向性;第二,对于结合位点分析,ChIP-Seq 通过寻找“峰”,结合分辨率可精确到10~30bp,而芯片上探针由于长度所限,无法精确定位,即使目前最高水平的商业芯片都无法提供可与ChIP-Seq媲美的分辨率;第三是所需样本数量。ChIP-chip需要多达4~5μg的起始样本,在杂交之前需要进行LM-PCR,但可能导致背景增高,竞争性扩增等导致假阳性。而ChIP-Seq仅需要纳克级起始材料,如SOLiD起始材料可低至20ng。两者技术特点如下: 研究方法CHIP-on-chip CHIP-Seq 分辨率30~100bp1bp 覆盖范围受芯片容量限制,只能选择性地扫 描特定区域,无法覆盖全基因组只要测定的序列(Reads)能够定位到基因组上,就能获得全部基因组信息 缺陷探针和非特异性区域杂交测序数据会有一些GC含量偏向 性价比只能研究在基因组上广泛存在的目 的位点(Broading bingding)可以扫描全基因组;可以研究在基因组上存在的稀有目的位点(Sharp bingding) 需要的DNA 量 高低(10~50bp)动态量程弱信号会被遗弃;强信号会饱和没有局限 选择数据产 出量 不可以可以
基因测序(PCR常见问题)
基因测序(PCR常见问题)生物专业很实用 PCR常见问题 PCR常见问题分析及对策(无扩增产物、非特异性扩增、拖尾、假阳性) 问题1:无扩增产物 现象:正对照有条带,而样品则无 原因: 1.模板:含有抑制物,含量低 2.Buffer对样品不合适 3.引物设计不当或者发生降解 4.反应条件:退火温度太高,延伸时间太短 对策: 1.纯化模板或者使用试剂盒提取模板DNA或加大模板的用量 2.更换Buffer或调整浓度 3.重新设计引物(避免链间二聚体和链内二级结构)或者换一管新引物 4.降低退火温度、延长延伸时间 问题2:非特异性扩增 现象:条带与预计的大小不一致或者非 特异性扩增带
原因: 1.引物特异性差 2.模板或引物浓度过高 3.酶量过多 4.Mg2+浓度偏高 5.退火温度偏低 6.循环次数过多 对策: 1.重新设计引物或者使用巢式PCR 2.适当降低模板或引物浓度 3.适当减少酶量 4.降低镁离子浓度 5.适当提高退火温度或使用二阶段温度法 6.减少循环次数 问题3:拖尾 现象:产物在凝胶上呈Smear状态。 原因: 1.模板不纯 2.Buffer不合适 3.退火温度偏低 4.酶量过多 5.dNTP、Mg 2+浓度偏高 6.循环次数过多 对策: 1.纯化模板 2.更换Buffer 3.适当提高退火温度 4.适量用酶 5.适当降低dNTP和镁离子的浓度 6.减少循环次数 问题4:假阳性 现象:空白对照出现目的扩增产物 原因: 靶序列或扩增产物 的交*污染 对策: 1.操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外; 2.除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管及加样枪头等均应一次性使用。 3.各种试剂最好先进行分装,然后低温贮存 PCR引物设计的黄金法则(转自tiangen)
全基因组重测序数据分析
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排 突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使 得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组 学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基 因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需 要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。
高通量测序生物信息学分析(内部极品资料,初学者必看)
基因组测序基础知识 ㈠De Novo测序也叫从头测序,是首次对一个物种的基因组进行测序,用生物信息学的分析方法对测序所得序列进行组装,从而获得该物种的基因组序列图谱。 目前国际上通用的基因组De Novo测序方法有三种: 1. 用Illumina Solexa GA IIx 测序仪直接测序; 2. 用Roche GS FLX Titanium直接完成全基因组测序; 3. 用ABI 3730 或Roche GS FLX Titanium测序,搭建骨架,再用Illumina Solexa GA IIx 进行深度测序,完成基因组拼接。 采用De Novo测序有助于研究者了解未知物种的个体全基因组序列、鉴定新基因组中全部的结构和功能元件,并且将这些信息在基因组水平上进行集成和展示、可以预测新的功能基因及进行比较基因组学研究,为后续的相关研究奠定基础。 实验流程: 公司服务内容 1.基本服务:DNA样品检测;测序文库构建;高通量测序;数据基本分析(Base calling,去接头, 去污染);序列组装达到精细图标准 2.定制服务:基因组注释及功能注释;比较基因组及分子进化分析,数据库搭建;基因组信息展 示平台搭建 1.基因组De Novo测序对DNA样品有什么要求?
(1) 对于细菌真菌,样品来源一定要单一菌落无污染,否则会严重影响测序结果的质量。基因组完整无降解(23 kb以上), OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;每次样品制备需要10 μg样品,如果需要多次制备样品,则需要样品总量=制备样品次数*10 μg。 (2) 对于植物,样品来源要求是黑暗无菌条件下培养的黄化苗或组培样品,最好为纯合或单倍体。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (3) 对于动物,样品来源应选用肌肉,血等脂肪含量少的部位,同一个体取样,最好为纯合。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (4) 基因组De Novo组装完毕后需要构建BAC或Fosmid文库进行测序验证,用于BAC 或Fosmid文库构建的样品需要保证跟De Novo测序样本同一来源。 2. De Novo有几种测序方式 目前3种测序技术 Roche 454,Solexa和ABI SOLID均有单端测序和双端测序两种方式。在基因组De Novo测序过程中,Roche 454的单端测序读长可以达到400 bp,经常用于基因组骨架的组装,而Solexa和ABI SOLID双端测序可以用于组装scaffolds和填补gap。下面以solexa 为例,对单端测序(Single-read)和双端测序(Paired-end和Mate-pair)进行介绍。Single-read、Paired-end和Mate-pair主要区别在测序文库的构建方法上。 单端测序(Single-read)首先将DNA样本进行片段化处理形成200-500bp的片段,引物序列连接到DNA片段的一端,然后末端加上接头,将片段固定在flow cell上生成DNA簇,上机测序单端读取序列(图1)。 Paired-end方法是指在构建待测DNA文库时在两端的接头上都加上测序引物结合位点,在第一轮测序完成后,去除第一轮测序的模板链,用对读测序模块(Paired-End Module)引导互补链在原位置再生和扩增,以达到第二轮测序所用的模板量,进行第二轮互补链的合成测序(图2)。 图1 Single-read文库构建方法图2 Paired-end文库构建方法
DNA测序结果中常见的几个问题
D N A测序结果中常见 的几个问题 公司内部档案编码:[OPPTR-OPPT28-OPPTL98-OPPNN08]
1 、为什么开始一段序列的信号很杂乱,几乎难以辨别 这主要是因为残存的染料单体造成的干扰峰所致,该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。 2 、为什么在序列的末端容易产生 N 值,峰图较杂 由于测序反应的信号是逐渐减弱的,所以序列末端的信号会很弱,峰图自然就会杂乱,加上测序胶的分辨率问题,如果碱基分不开,就会产生N 值,正常情况下ABI377测序仪能正确读出500个碱基的有效序列。 3 、测序结果怎么找不到我的引物序列 如果找不到测序所用的引物序列。这是正常的,因为引物本身是不被标记的,所以在测序报告中是找不到的;如果找不到克隆片段中的扩增引物,可能是您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到扩增引物;另外插入片段的插入方向如果是反的,此时需找引物的互补序列。 4 、测序结果怎么看不到我克隆的酶切位点 可能的原因同上,您克隆的酶切位点距离您的测序引物太近,开始一段序列很杂,几乎难以辨别,有可能看不清或看不到酶切位点。通常我们会尽量选择距离酶切位点远点的引物,当然,若是样品出现意外原因,如空载、载体自连等,克隆的酶切位点也是看不到的。 5 、你测出的结果与我预想的不一致,给我的结果与我需要的序列有差距,这是怎么回事
首先,我们会核实给您的测序结果是否对应您的样品编号,如果对应的是您的样品,由于不知您的实验背景,测得的序列是否与您预想的结果一致我们无法判断,我们能做到的是检查发送给您的测序结果和您提供来的样品是否一致。 6 、序列图为什么会有背景噪音(杂带)是否会影响测序结果 序列图的背景杂带是由荧光染料引起,如果太强会影响测序结果,要看信噪比,我们给的结果信噪比大都在98%以上。 7 、测序结果为什么与标准序列有差别 原因可能有:样品个体之间的差别、测序准确率的问题,自动测序仪分析序列的准确并非100%,建议至少测一次双向,通过双向测序可以最大限度减少测序的错误。当然尽管我们有时做了最大努力,但还是保证不了和文献序列完全一致,但我们测序报告是客户样品序列的真实结果。 8 、 PCR 产物测序与克隆后测序序列为什么有差别 PCR 产物克隆到载体中进行测序,有两个方面可能序列有变化:首先,PCR 扩增过程中可能产生错配。将片段克隆到载体中也有可能发生突变;其次,测序的准确率并非100%。 9 、有杂合位点,但你们的报告上看不到杂合的信号! 如果在您认为应该出现杂合信号的位置上只出现单一的信号,那么可能是您样品突变的模板与正常的模板的比例没达到可以测出的浓度。测序反应的信号强度直接与模板的量有关,如果突变的模板所占的比例很低,仪器会自动将它作为背景信号了,很难检测出来。只有当测序反应体系中正常的和突变的模板量比较接近时,才能较可靠地检测到突变体
利用SeqMan进行序列拼接
利用SeqMan进行序列拼接 Step1:打开Seqman软件 Step2:加入你要拼接的序列 点击Add sequences 查找并选中要拼接的序列(可按住control键进行多选) 点击Add按钮填加选择的序列 填加完后点击done 注:最好用测序的图谱尽量不要直接用测序得到的序列 Step3:去除末端序列 主要是去除序列末端测序质量差或是载体序列 有两种方法可以用来去除这类末端序列 其一:利用Seqman自带的去除工具自动去除(利用Trim ends按钮进行) 其二:手工去除 个人感觉手工去除方法最有效,因此下边我们以后工去除为例进行演示 手工去除侧翼序列 双击要去除侧翼序列的目标序列 将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标会变成一个有两个箭头的水平线按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变浅,这部分变浅的序列则就被去除,不再参加后面的拼接
此步请将测序不准确或认为是载体的序列用这种方法去除。 测序准确的峰形图 峰形规则,一般在序列的中部,如下图所示 测序不准确的峰形图 峰形较乱,很难判断是哪个碱基,一般位于序列两端,如下图所示
Step4:进行序列拼接 点击Assemble按钮 在新出现窗口处点击拼接好的contig1 在出现的Alignment of contig1 窗口中点击左三角显示序列的测序图谱点击菜单contig->strategy view可以观察序列拼接的宏观图 Step5:查找拼接错误 find conflict 点击菜单Edit 点击Find Previous或Find Next查找接接中出现的错误 还可以通过Seqman左下角的快捷按钮查找错误的拼接
测序过程常见问题分析与解答
测序过程常见问题分析与解答 1、DNA测序样品用什么溶液溶解比较好? 答:溶解DNA测序样品时,用灭菌蒸馏水溶解最好。DNA的测序反应也是Taq酶的聚合反应,需要一个最佳的酶反应条件。如果DNA用缓冲液溶解后,在进行了测序反应时,DNA溶液中的缓冲液组份会影响测序反应的体系条件,造成Taq酶的聚合性能下降。有很多客户在溶解DNA测序样品时使用TE Buffer。的确,TE Buffer能增加DNA样品保存期间的稳定性,但TE Buffer对DNA测序反应有影响,根据我们的经验,我们还是推荐使用灭菌蒸馏水来溶解DNA测序样品。 2、提供DNA测序样品时,提供何种形态的比较好? 答:我们推荐客户提供菌体,由我们来提取质粒,这样DNA样品比较稳定。如果您要以提供DNA样品,我们也很欢迎,但一定要注意样品纯度和数量。提供的测序样品为PCR产物时,特别需要注意DNA的纯度和数量。PCR产物应该进行切胶回收,否则无法得到良好的测序效果。有关DNA测序样品的详细情况请严格参照“测序模板的要求”部分的说明。 3、提供的测序样品为菌体时,以什么形态提供为好? 答:一般菌体的形态有:平板培养菌、穿刺培养菌,甘油保存菌或新鲜菌液等。我们提倡寄送穿刺培养菌或新鲜菌液。平板培养菌运送特别不方便,我们收到的一些平板培养菌的培养皿在运送过程中常常已经破碎,面目全非,需要用户重新寄样。这样既误时间,又浪费客户的样品。一旦是客户非常重要的样品时,其后果更不可设想。而甘油保存菌则容易污染。制作穿刺菌时,可在1.5ml的Tube管中加入琼脂培养基,把菌体用牙签穿刺于琼脂培养基(固体)中,37℃培养一个晚上后便可使用。穿刺培养菌在4℃下可保存数个月,并且不容易污染,便于运送。 4、与测序引物有关的问题