透射电镜超薄切片步骤
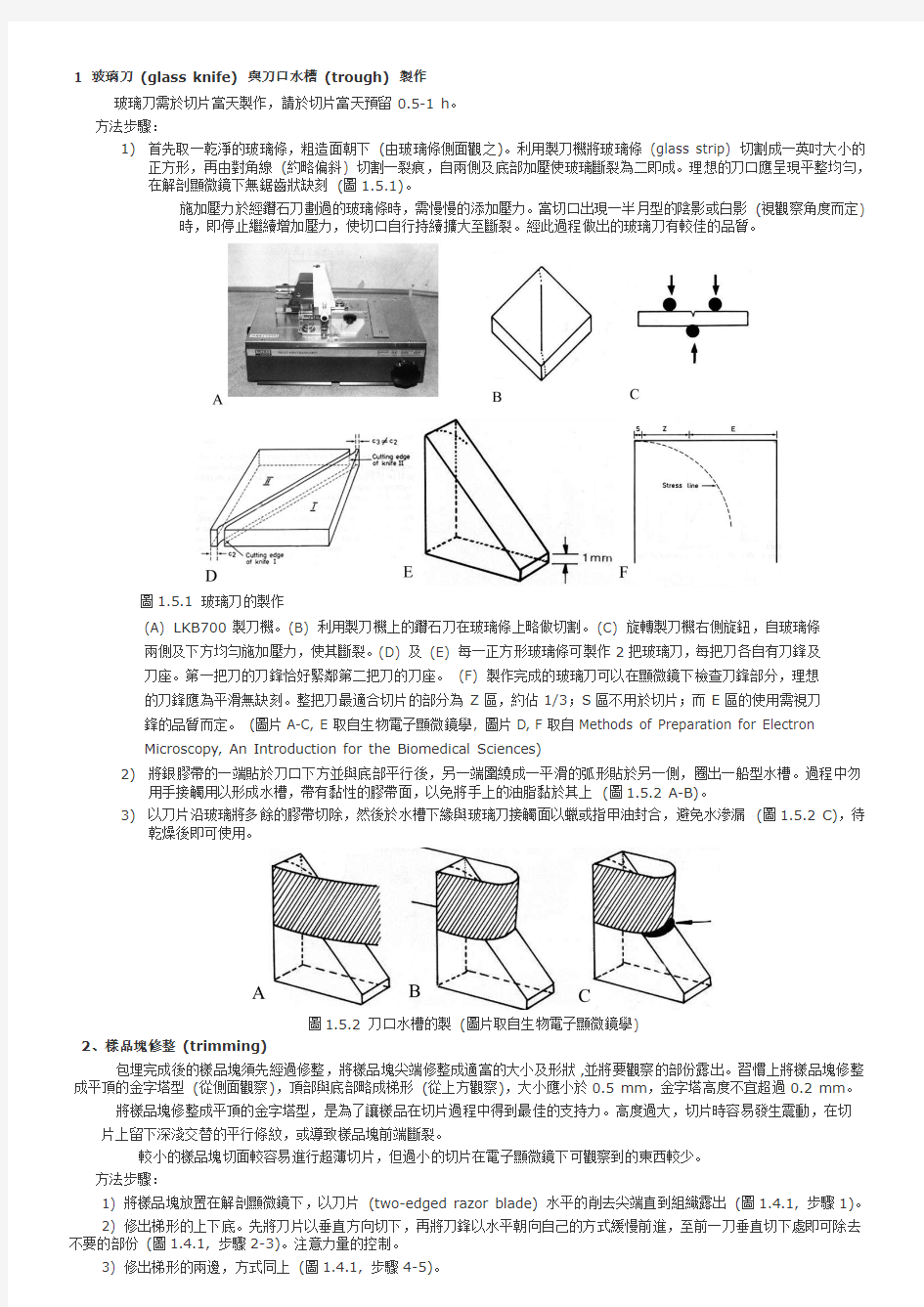

1 玻璃刀(glass knife) 與刀口水槽(trough) 製作
玻璃刀需於切片當天製作,請於切片當天預留0.5-1 h。
方法步驟:
1)首先取一乾淨的玻璃條,粗造面朝下(由玻璃條側面觀之)。利用製刀機將玻璃條(glass strip) 切割成一英吋大小的
正方形,再由對角線(約略偏斜) 切割一裂痕,自兩側及底部加壓使玻璃斷裂為二即成。理想的刀口應呈現平整均勻,在解剖顯微鏡下無鋸齒狀缺刻(圖1.5.1)。
施加壓力於經鑽石刀劃過的玻璃條時,需慢慢的添加壓力。當切口出現一半月型的陰影或白影(視觀察角度而定)
時,即停止繼續增加壓力,使切口自行持續擴大至斷裂。經此過程做出的玻璃刀有較佳的品質。
圖1.5.1 玻璃刀的製作
(A) LKB700製刀機。(B) 利用製刀機上的鑽石刀在玻璃條上略做切割。(C) 旋轉製刀機右側旋鈕,自玻璃條
兩側及下方均勻施加壓力,使其斷裂。(D) 及(E) 每一正方形玻璃條可製作2把玻璃刀,每把刀各自有刀鋒及
刀座。第一把刀的刀鋒恰好緊鄰第二把刀的刀座。(F) 製作完成的玻璃刀可以在顯微鏡下檢查刀鋒部分,理想
的刀鋒應為平滑無缺刻。整把刀最適合切片的部分為Z區,約佔1/3;S區不用於切片;而E區的使用需視刀
鋒的品質而定。(圖片A-C, E取自生物電子顯微鏡學, 圖片D, F取自Methods of Preparation for Electron
Microscopy, An Introduction for the Biomedical Sciences)
2)將銀膠帶的一端貼於刀口下方並與底部平行後,另一端圍繞成一平滑的弧形貼於另一側,圈出一船型水槽。過程中勿
用手接觸用以形成水槽,帶有黏性的膠帶面,以免將手上的油脂黏於其上(圖1.5.2 A-B)。
3)以刀片沿玻璃將多餘的膠帶切除,然後於水槽下緣與玻璃刀接觸面以蠟或指甲油封合,避免水滲漏(圖1.5.2 C),待
乾燥後即可使用。
圖1.5.2 刀口水槽的製(圖片取自生物電子顯微鏡學)
2、樣品塊修整(trimming)
包埋完成後的樣品塊須先經過修整,將樣品塊尖端修整成適當的大小及形狀,並將要觀察的部份露出。習慣上將樣品塊修整成平頂的金字塔型(從側面觀察),頂部與底部略成梯形(從上方觀察),大小應小於0.5 mm,金字塔高度不宜超過0.2 mm。
將樣品塊修整成平頂的金字塔型,是為了讓樣品在切片過程中得到最佳的支持力。高度過大,切片時容易發生震動,在切片上留下深淺交替的平行條紋,或導致樣品塊前端斷裂。
較小的樣品塊切面較容易進行超薄切片,但過小的切片在電子顯微鏡下可觀察到的東西較少。
方法步驟:
1) 將樣品塊放置在解剖顯微鏡下,以刀片(two-edged razor blade) 水平的削去尖端直到組織露出(圖1.4.1, 步驟1)。
2) 修出梯形的上下底。先將刀片以垂直方向切下,再將刀鋒以水平朝向自己的方式緩慢前進,至前一刀垂直切下處即可除去不要的部份(圖1.4.1, 步驟2-3)。注意力量的控制。
3) 修出梯形的兩邊,方式同上(圖1.4.1, 步驟4-5)。
4) 待初步形狀出來後,換一新刀片再做更細微的修整,盡量使上下底平行,切面光滑無缺刻。此步驟會影響連續切片(ribbon) 的形成。
圖 1.4.1 樣品塊修整(圖片取自
Methods of Preparation for Electron
Microscopy)
3、半薄切片(1-2um)、光学显微镜定位
4、刀槽及液面的调节
超薄切片只有漂浮在水面上或其他合适的液面上才便于收集、伸展和捞取,为此在刀口背部装上一个小的水容器,通常称为“刀槽”。通常用定制的硬塑料刀槽架或用一小段医用橡皮胶缠绕过刀背,紧紧地粘在玻璃上,橡皮胶的后下部分用融化的牙科用红腊密封,以防漏水。注意橡皮胶的顶端不应高出刀刃。切片前在刀槽内加入双蒸水,使切后的切片漂浮于液面上。液面的调节一般先加稍过量的液体使之凸起,以保证浸湿刀刃。然后在双筒解剖镜下用注射器或移液管慢慢吸去一部分液体,调整照明系统,当液面变成凹面、可见一反射光线的弧面,即为正确的液面。从这一光亮的反射光可看到切出的片子。在切片过程中由于液体蒸发,需加液以保证正确的液面。
5、超薄切片
切片时将夹着修好组织块的样品夹安装在切片机的样品臂上,刀台上装好玻璃刀,其高度要与刀台上的标准规等高,后斜角一般在3~5°,在显微镜下使样品块面的上下两条平行边与刀刃平行,如是梯形的应长边在下面。调整好刀刃和组织块的距离,再在刀槽内注入适量的双蒸馏水。将切速放在2~5mm/秒,调节控制切片厚度的电流表,即可切片。当切片停止时,立即打开电吹风,冷却样品臂。捞片后关掉电吹风又可重新切片,或更换新刀。
切片厚度的判断。一般是利用反射光干涉色来判断切片的厚度,即从切片表面反射的光和从切片下面的水面的反射光发生干涉现象(见下表)。当用白光时,从双筒显微镜下观察两种光线间的差别。
目前公认的树脂包埋切片厚度与干涉色的近似值
干涉色切片厚度
暗灰色40nm以下
灰色40~50nm
银白色50~70nm
金黄色70~90nm
紫色90nm以上
紫色切片太厚,电子束不易穿过,故不适用于电镜观察用。金黄色切片其分辨率也比较低。目前一般认为银白色的切片是较理想的厚度,其分辨率可达2.5nm左右。也有人喜欢采用金黄色切片,尤其在细胞化学、放射自显影中较适宜。
切片的展平和捞取。在切削过程中,由于切片很薄,经挤压使切片发皱,甚至引起标本结构变形。为此,常用沾有氯仿或二甲苯的滤纸小片移近刀槽液面,试剂挥发性蒸汽可使切片软化伸展。蒸发时间2~3秒钟即可,不能把试剂滴入水槽。
捞片时,可用眼睫毛做成的小针把切片轻轻地拨离刀刃,并按需要把切片带分成几段。再用弯头的小镊子夹住铜网的边缘,有支持膜的一面向下,与切片轻轻接触,使切片贴附在铜网的膜上,轻轻提起并把膜面朝上,放于培养皿中无毛的滤纸上,待干经染色后即可电镜观察。
方法步驟:
1) 將標本臂(specimen arm) 及刀座(knife holder) 退回起點。
標本臂的控制懸鈕位於切片機右後側面,退標本臂至底時先以順時鐘方向旋轉至底,再以反方向倒轉半圈。
2) 選用適當的固定座將樣品塊固定於標本臂上,加以旋緊。略為調整樣品塊的角度(圖1.6.2 中3,4),使樣品塊整個切
面與即將放置的刀口等距平行。緊量使其在固定過程中勿大力震動標本臂,以免影響儀器準確度。
3) 將玻璃刀安置在刀座上,調整適當的高度與位置,並加以旋緊
固定不良的樣品塊與玻璃刀可能在切片上造成chatter的現象。
4) 調整玻璃刀的傾斜角度(clearance angle) 為4° (圖1.6.3中7)。
此角度視包埋劑與刀子種類的不同而改變,但一般都在2~5°間。
5) 鬆開刀座,以手動方式逐漸前移刀座,至刀口靠進樣品塊時鎖緊刀座 (圖1.6.3中1左扳時可用手扶住整個刀座,慢
慢向前推進。右扳時可固定鎖緊刀座)。
1
6
789
9
圖1.6.3 Richert-jung Ultracut E 超薄切片機刀座
圖上各部位說明:(1) 刀座固定-右扳可固定刀座。 (6) 鎖緊玻璃刀於刀座上。(7) 調整玻璃刀的傾斜角度
(clearance angle)。(8) 在1右扳情況下,順時鐘方向旋轉可前進刀座;逆時鐘方向旋轉可後退刀座 (圖片取
自生物電子顯微鏡學)。(9) 左右調整刀座角度。
6) 利用注射針筒在水槽中加蒸餾水,加至水面在顯微鏡下剛好呈一均勻亮面 (圖1.6.4 C)。
不正確的水面高度可由水槽中出現黑影得知。水面過低時,多在刀口附近水面形成黑色弧線,此時剛切出的切片可能黏附在刀口上,並擠壓皺縮 (圖1.6.4 A)。 水面過高時,水面變得灰暗,可能導致樣品塊沾溼,剛切出的切片被拖離水槽 (圖1.6.4 B)。
圖1.6.4 刀口水槽的水面高度與切片的關係
(圖片取自Principles and Techniques of Electron Microscopy, Biological Applications Volume 1)
7) 將玻璃刀朝樣品塊逼近,至快接近時停止,需小心勿使兩者碰撞 (圖1.6.3中8)。
8) 選擇修平樣品塊切面與切厚切片所用的刀鋒 (圖1.6.1)。
9) 在顯微鏡下調整玻璃刀刀鋒與樣品塊的距離與角度 (圖1.6.5)。此部分難度較高,利用反射的亮帶判斷距離需經驗和
適當光源的配合。需小心勿使刀鋒超過樣品塊平面,否則整個梯形都可能斷裂。
使樣品塊梯形的上下底與刀口平行:梯形切面上下底與刀口不平行時,旋轉樣品塊使之平行 (圖1.6.2中3,4)。 使樣品塊整個梯形平面與刀口等距:(1) 水平面等距-樣品塊切面上之光帶兩端寬度不一時,代表刀口與樣品面
互相歪斜,旋轉刀座,使光帶寬度一致 (圖1.6.3中9)。(2) 垂直面等距-樣品塊切面上之光帶寬度會隨標本臂上下而改變,代表梯形之上下底與刀口距離不同,旋轉樣品塊角度,使光帶寬度一致 (圖1.6.2中3,4)。
圖1.6.5 刀口與組織切面的調整
A樣品塊整個梯形平面與刀口保持垂直面等距。B樣品塊整個梯形平面與刀口保持水平面等距。C樣品塊梯形的上下底與刀口平行。(圖片取自Methods of Preparation for Electron Microscopy, An Introduction for the Biomedical Sciences)
10) 調整切片速度為3 mm/sec及semisection切片厚度1 μm,在進行超薄切片(ultrasection)前需先進行厚片
(semisection) 的觀察及樣品塊切面的微修整。
11) 調整標本臂開始切片的起始位置(樣品塊略低於刀鋒)。極重要,若無法確定,請一定要請教有經驗的老師。
12) 開始切厚片,待切出一平面時,可以單面削平的竹籤撈起觀察。方式如下:在玻片上滴一滴水,以撈起切片的竹籤尖
端接觸玻片上的水後,將玻片放置在55~60℃加熱板上待其乾燥。滴一滴toluidine blue在切片上,同樣在55~60℃加熱板上加熱約30 sec (此時染劑邊緣會出現金色光澤),再以洗瓶沖洗掉過多的染劑後放回加熱板,當玻片乾燥後即可放在顯微鏡下觀察。
13) 在厚片上已切到所欲觀察的目標,且已切出完整的切面時,即可開始切超薄切片。調整切片速度為3 mm/sec及
ultrasection切片厚度為70~90 nm。做免疫電顯實驗的切片須較厚,可定為99 nm。
14) 觀察刀鋒是否有毀損。若有,須將刀座稍微退離樣品塊(圖1.6.3中8) 後換一刀鋒,並重新對刀,方法同步驟7-9。
15) 切出來的切片在顯微鏡下應呈銀白色(60~90 nm) 至金黃色(90~150 nm),直線前進的連續切片帶(ribbon)。
做免疫電顯實驗的切片應呈金黃色。
14) 待切出適當長度的切片帶後停止切片。以尖端黏有眉毛的竹籤輕划水面,將切片待趕至水槽深處,並以眉毛筆輕觸切
片帶邊緣缺刻部位,使切片帶斷至適當長度(約5, 6片)。
15) 以鑷子夾取鎳網(Ni grid,免疫電顯實驗需用鎳網) 邊緣來撈取切片。撈取切片的方式有二:(1)將鎳網欲接觸切片
的那一面朝上傾斜進入水中,以上緣接觸第一片切片後緩緩拉起。(2) 將鎳網欲接觸切片的那一面朝下,由上方向下覆蓋切片。
買來的grid有兩面,在燈光下顏色較淡且不反光的稱為dull side,顏色較深且邊緣有明顯反光的稱為shining side。未免將來混淆,應固定使用同一面。
16) 將撈到切片的grid放在濾紙上或以濾紙從未黏附切片的一面接觸grid,以濾紙吸乾水分後放入保存盒內,打開盒蓋,
置於電子乾燥箱中,待完全乾燥後(1~2 d) 蓋上盒蓋保存。
切片一定須等到完全乾燥後方可用電子顯微鏡觀察。
透射电镜样品制备方法
透射电镜样品制备方法 由于电子束穿透能力限制,必须把标本切成厚度小于0.1um以下的薄片才适用,这种薄片称为超薄切片。常用的超薄切片厚度是50-70nm。 在透射电镜的样品制备方法中,超薄切片技术是最基本、最常用的制备技术。超薄切片的制作过程基本上和石蜡切片相似,需要经过取材、固定、脱水、渗透、包埋聚合、切片及染色等步骤。 一.取材的基本要求 组织从生物活体取下后,如果不立即进行适当处理,会由于细胞内部的各种酶作用,出现细胞自溶现象。此外还可能由于污染,微生物在组织内繁殖使细胞的微细结构遭受破坏,因此,为了细胞结构尽可能保持天然状态,必须做到快、小、准、冷。 (1)动作迅速,组织从活体取下后应在最短的时间内(争取1分钟内)投入2.5%戊二醛固定液。 (2)所取组织的体积要小,一般不超过1mm*1mm*1mm。也可将组织修成1mm*1mm*2mm大小长条形。因为固定剂的渗 透能力较弱,组织块如果太大,块的内部将不能得到良好的 固定。 (3)机械损伤要小,解剖器械应锋利,操作宜轻,避免牵拉、挫伤与挤压。 (4)操作最好在低温(0℃~4℃)下进行,以降低酶的活性,防
止细胞自溶。 (5)取材部位要准确。 二.取材方法 将取出的组织放在洁净的蜡版上,滴一滴预冷的固定液,用新的、锋利的刀片将组织切下并修小,然后用牙签或者镊子将组织块移至盛有冷的固定液的1.5ml离心管中。如果组织块带有较多的血液和组织液,应先用PBS洗几遍,然后切成小块固定。 1.动物及人体组织的取材(在冰浴上进行) 动物组织的取材,应麻醉(1%戊巴比铵5ml/kg体重腹腔注射)或断头急性处死,解剖出所需器官,用解剖剪刀剪取一 小块组织,放在干净的纸板上,滴一滴冷却的固定液,用新的、无油污的锋利双面刀片将材料切成大约1mm宽,2~3mm长的 小块并从中选出受损伤较小的小条,再将其切成1mm3的小块,最后用牙签将这些小块逐一放入盛有预冷的、新鲜固定液的 1.5ml管内,放入冰箱冷藏室低温固定(0~4℃)2-4小时或以 上。 *固定结束后,将固定液用PBS稀释三倍,样品于该溶液中在4℃冰箱保存。送样前请用PBS浸泡清洗3次,贴好标签 送至电镜室。 2.体外培养细胞的取材(在冰浴上进行) 培养在培养瓶中的细胞取材时,先倒出部分培养液,然后用刮刀轻轻刮下瓶壁上的细胞,将细胞悬液转移到离心管中,
透射电镜的样品制备方法详解
透射电镜的样品制备 透射电镜的样品制备是一项较复杂的技术,它对能否得到好的TEM像或衍射谱是至关重要的.投射电镜是利用样品对如射电子的散射能力的差异而形成衬度的,这要求制备出对电子束"透明"的样品,并要求保持高的分辨率和不失真.电子束穿透固体样品的能力主要取决加速电压,样品的厚度以及物质的原子序数.一般来说,加速电压愈高,原子序数愈低,电子束可穿透的样品厚度就愈大.对于100~200KV的透射电镜,要求样品的厚度为50~100nm,做透射电镜高分辨率,样品厚度要求约15nm(越薄越好). 透射电镜样品可分为:粉末样品,薄膜样品,金属试样的表面复型.不同的样品有不同的制备手段,下面分别介绍各种样品的制备. (1)粉末样品因为透射电镜样品的厚度一般要求在100nm以下,如果样品厚于100nm,则先要用研钵把样品的尺寸磨到100nm以下,然后将粉末样品溶解在无水乙醇中,用超声分散的方法将样品尽量分散,然后用支持网捞起即可. (2)薄膜样品绝大多数的TEM样品是薄膜样品,薄膜样品可做静态观察,如金相组织;析出相形态;分布,结构及与基体取向关系,错位类型,分布,密度等;也可以做动态原位观察,如相变,形变,位错运动及其相互作用.制备薄膜样品分四个步骤: a将样品切成薄片(厚度100~200微米),对韧性材料(如金属),用线锯将样品割成小于200微米的薄片;对脆性材料(如Si,GaAs,NaCl,MgO)可以刀将其解理或用金刚石圆盘锯将其切割,或用超薄切片法直
接切割. b切割成φ3mm的圆片用超声钻或puncher将φ3mm薄圆片从材料薄片上切下来. c预减薄使用凹坑减薄仪可将薄圆片磨至10μm厚.用研磨机磨(或使用砂纸),可磨至几十μm. d终减薄对于导电的样品如金属,采用电解抛光减薄,这方法速度快,没有机械损伤,但可能改变样品表面的电子状态,使用的化学试剂可能对身体有害.对非导电的样品如陶瓷,采用离子减薄,用离子轰击样品表面,使样品材料溅射出来,以达到减薄的目的.离子减薄要调整电压,角度,选用适合的参数,选得好,减薄速度快.离子减薄会产生热,使样品温度升至100~300度,故最好用液氮冷却样品.样品冷却对不耐高温的材料是非常重要的,否则材料会发生相变,样品冷却还可以减少污染和表面损伤.离子减薄是一种普适的减薄方法,可用于陶瓷,复合物,半导体,合金,界面样品,甚至纤维和粉末样品也可以离子减薄(把他们用树脂拌合后,装入φ3mm金属管,切片后,再离子减薄).也可以聚集离子术(FIB)对指定区域做离子减薄,但FIB很贵.对于软的生物和高分子样品,可用超薄切片方法将样品切成小于100nm的薄膜.这种技术的特点是样品不会改变,缺点是会引进形变.(3)金属试样的表面复型即把准备观察的试样的表面形貌(表面显微组织浮凸)用适宜的非晶薄膜复制下来,然后对这个复制膜(叫做复型)进行透射电镜观察与分析.复型适用于金相组织,断口形貌,形变条纹,磨损表面,第二相形态及分布,萃取和结构分析等. 制备复型的材料本身必须是"无结构"的,即要求复型材料在高倍成像时也
电镜切片样品制作步骤知识讲解
A 悬浮培养的细胞、细菌、血细胞、精子等; 细胞使用PBS或无血清培养基离心漂洗1~2次以去除血清,离心转速依据不同离心机、不同样品自定,总时间控制在5min内;细胞团根据预设浓度在适量2.5%戊二醛吹悬,滴加在预先置入青霉素小瓶中的托盘,4℃静置沉降2~3天,在托盘周围加入PBS,以防止样品干燥。 B贴壁培养的细胞: 在培养皿中预先加入盖玻片,使细胞贴附于盖玻片上;PBS或无需请培养基漂洗后,放置在培养板室温固定1h,4℃3 h,注意放置干燥,自行转入青霉素小瓶中,加满PBS送检。SEM标本处理必须使用玻璃容器,需明确所用盖玻片尺寸可以放入青霉素瓶。 C组织取材 样品观察表面可达8~10mm2,高度小于5mm左右; 样品表面在固定前必须清洁:使用生理盐水或PBS冲洗掉表面的灰尘以及不需要观察的蛋白、粘液等。能够明确标识标本的观察面。消化道、呼吸道、血管、生殖器官、泌尿等官腔内表面,尤其要注意先清洗再固定。 如需要观察脏器内结构,应依照不同的实验目的,决定目的脏器是否需要灌注清洗; 固定2.5%戊二醛浸没标本,室温1h,4℃固定3h以上,换PBS送检。 附:2.5%戊二醛的配制 Step 1: 0.2M磷酸缓冲液的配制: --------------------- 磷酸二氢钠(NaH2PO4.H2O) 2.6克 磷酸氢二钠(Na2HPO4.12H2O)29克 双蒸馏水加至500毫升 pH调至7.4 Step 2: 戊二醛固定液的配制: --------------------- 25% 戊二醛1ml 双蒸馏水4ml 0.2mol/L磷酸缓冲液5ml 戊二醛最终浓度 2.5% pH值7.3-7.4
透射电镜样品制作(2015-12-11)
透射电镜样品包埋块制作原理步骤 一、取材: 1、动作迅速:组织离体后,应将其快速放入4℃戊二醇固定液中,使组织细胞尽可能保持原来的生活状态。 2、减少损伤:选择锋利切割器械,减少牵拉或挤压组织。 3、组织块大小:取材医生可先切成长条形,然后再修成约1mm3大小。 二、固定: 固定目的是把细胞在活体状态时的超微结构细节尽可能完整地保存下来,避免自身酶的分解而出现自溶,或因外界微生物的入侵繁殖而产生腐败,致使细胞的超微结构遭受破坏。同时也使细胞内的各种成分固定下来,避免以后的冲洗和脱水时溶解和流失。理想的固定剂应具备以下特点:能够迅速又均匀地渗透到组织结构内;能够稳定细胞各种结构成分,使之在以后处理过程中不致溶解和丢失;对细胞超微结构没有损伤;能供细胞化学测定并能增强图像反差。当然,满足所有要求的固定剂是不存在的,目前常用固定剂有锇酸和戊二醇。 1.使用锇酸的注意事项: 锇酸即四氧化锇,它能和细胞内绝大多部分成分反应,且能够保护脂肪,但对碳水化合物、糖类和核酸保护作用差,锇酸渗透差,分子密度大,经锇酸固定的组织在电镜下能获得较好反差。锇酸为剧毒、极易挥发的试剂,对呼吸道有强烈刺激作用,必须在通风橱中操作,废液必须收集在密闭容器中。常用的锇酸溶液为2%的储备液,使用之前需用0.2MPBS稀释成1%的锇酸溶液。 2.戊二醇 戊二醇能够稳定糖原,同时保存某些锇酸保护作用差的蛋白质结构,对酶活性破坏小。对微管、滑面内质网等固定较好,对脂肪保护差,且反差小,因此必须和锇酸配合使用,即“双重固定法”。 3.固定方法: 样本先用2.5%戊二醇在4℃下固定2h,经PBS缓冲液多次清洗后再用1%锇酸固定2h。根据不同组织,可适当延长固定时间。由于戊二醇能够和锇酸反应产生电子致密的还原锇沉淀,组织经戊二醇固定后,必须将戊二醇清洗干净才能转入锇酸。此外,锇酸又能和乙醇作用生成沉淀,因此锇酸固定后也应用PBS清洗液进行清洗干净方能进行脱水处理。一般清洗3次,每次15min左右(或过夜)。 三、脱水:脱水是将组织中的游离水彻底清除的过程。由于常用的包埋剂,如环氧树脂,大多都是非水溶 性树脂,只有将生物组织中的游离水清除干净,包埋剂才能浸入组织,常用脱水剂是乙醇和丙酮。乙醇对细胞物质抽提少,组织收缩也少,但它和环氧树脂互溶性差,因此使用乙醇脱水时须用环氧丙烷作为中间溶剂。丙酮和酒精、环氧丙烷互溶,所以通常先用乙醇后再用丙酮的脱水方法。急剧脱水会引起细胞收缩,因此应采用逐级脱水(50%-70%-90%-100%乙醇)而不能急剧脱水。更换溶液时动作要快,特别是不要让组织离开溶液,否则会在组织内外产生气泡;脱水过程中若要长时间停留或过夜,应放在70%乙醇或丙酮中,并在4℃保存。 四、包埋 1.渗透:渗透就是用包埋剂逐渐取代组织中的脱水剂,使细胞内外空隙被包埋剂所填充。一般可先用环氧丙烷对半稀释的包埋剂浸透1-2h,再用纯包埋剂37℃烤箱渗透2h左右。包埋剂通常由树脂、硬化剂、增塑剂及催化剂4种试剂按一定比例配制而成。 2.包埋:目的是以包埋剂完全浸透到组织内部,经加温逐渐聚合成坚硬固体。理想包埋剂应具备的条件:黏稠适中,有良好切割性;能经受电子轰击;透明度较好;对人体无害。目前常用国产环氧树脂618、Epon812环氧树脂、及低黏度包埋剂Spurr。 3.环氧树脂:环氧树脂为热塑性树脂,主要有两种化学反应基团,即环氧基和羟基。末端环氧基易与其他含活性氢原子化合物如胺类反应,形成首尾相接的长链状聚合物。单体中羟基能与酸酐结合,形成分子间横桥连接。因此,把环氧树脂单体、胺类、酸酐等三者按一定比例混合,加上适当温度,可形成稳定的交 1
电镜切片样品制作步骤
扫描电镜样品的准备 A悬浮培养的细胞、细菌、血细胞、精子等; 细胞使用PBS或无血清培养基离心漂洗1~2次以去除血清,离心转速依据不同离心机、不同样品自定,总时间控制在5min内;细胞团根据预设浓度在适量 2.5%戊二醛吹悬,滴加在预先置入青霉素小瓶中的托盘,4℃静置沉降2~3天,在托盘周围加入PBS,以防止样品干燥。 B贴壁培养的细胞: 在培养皿中预先加入盖玻片,使细胞贴附于盖玻片上;PBS或无需请培养基漂洗后,放置在培养板室温固定1h,4℃3 h,注意放置干燥,自行转入青霉素小瓶中,加满PBS送检。 SEM标本处理必须使用玻璃容器,需明确所用盖玻片尺寸可以放入青霉素瓶。 C组织取材 样品观察表面可达8~10mm2,高度小于5mm左右; 样品表面在固定前必须清洁: 使用生理盐水或PBS冲洗掉表面的灰尘以及不需要观察的蛋白、粘液等。能够明确标识标本的观察面。消化道、呼吸道、血管、生殖器官、泌尿等官腔内表面,尤其要注意先清洗再固定。 如需要观察脏器内结构,应依照不同的实验目的,决定目的脏器是否需要灌注清洗;固定 2.5%戊二醛浸没标本,室温1h,4℃固定3h以上,换PBS送检。 附: 2.5%戊二醛的配制
Step 1: 0.2M磷酸缓冲液的配制:--------------------- 磷酸二氢钠(NaH2PO 4.H2O) 2.6xx 磷酸氢二钠(Na2HPO 4.12H2O)29xx 双蒸馏水加至500毫升pH调至 7.4 Step 2: 戊二醛固定液的配制: --------------------- 25%戊二醛1ml 双蒸馏水4ml 0.2mol/L磷酸缓冲液5ml 戊二醛最终浓度 2.5% pH值 7.3-
透射电镜常规样品制备流程
透射电镜样品制备流程 由于透射电镜能观察的样品必须很薄(60~70nm),所以透射电镜的样品准备要求很严格,方法也很单一,仅有一下两种方法: 一.负染色技术 负染色技术简单快速,可以显示生物大分子、细菌、分离的细胞器以及蛋白晶体等样品的形态、结构、大小以及表面结构的特征。尤其在病毒学中,负染色技术有着广泛的应用。 样品要求:①样品悬液的纯度不要求很纯,但是如果杂质太多,如大量的细胞碎片,培养基残渣,糖类以及各种盐类结晶的存在都会干扰染色反应和电镜的观察。尤其是不能有过多的糖类,因为在电子束的轰击下,糖类容易碳化而有碍观察,因此样品要适当提纯。②样品悬液的浓度要适中,太稀在电镜下很难找到样品,太浓样品堆积影响观察。 操作流程:吸取样品悬液滴到有膜的铜网上,静置数分钟,然后用滤纸吸去多余的液体,滴上负染色液,染色1~2min后滤纸吸去负染色液,待干后用于电镜观察。 二、超薄切片技术 超薄切片技术是为透射电镜观察提供薄样品的专门技术,是生物学中研究细胞超微结构最常用的技术。广泛应用于生物体的各种细胞的超微结构观察。一般厚度在10~100nm的切片称为超薄切片,制作这种切片的技术叫做超薄切片技术。超薄切片制作的过程包括取材、固定、脱水、渗透、包埋、聚合、切片和染色等几个环节,和一般光学显微镜的石蜡切片过程相似。但是,超薄切片切片过程更为细致与复杂,要求更严格,而且所用的试剂比较昂贵、配制复杂、强致癌。具体操作步骤、注意事项如下: 1.取材和前固定:快速的切取大小为0.5~1.0mm3的样品块,一分钟内把组织(样品)块浸入2.5%戊二醛(进口品质)溶液(取样前来平台领取),每个离心管内装20个以上的样品块,作为一个样送到平台。要求:①取材前一定要和工作人员取得电话联系!②取材选择部位要准确可靠,确保每块材料都是要观察的部位。③所有植物样品一定要抽真空,能够沉底的样品也抽真空15mins,不能沉底的样品一定要抽真空致沉底!④细菌、散在细胞等不能成块的样品,加戊二醛固定液,离心沉淀后送到平台,由平台工作人员处理。⑤泡在前固定液的材料最多可以放2周。 2.漂洗:用1M PBS Buffer Ph 7.2冲洗3次,每次15mins。 3.后固定:加入1%锇酸处理到样品变黑。植物样品一般处理2.5hours,真菌、细菌一般处理2hours,动物样品处理1hours。注意事项:①锇酸剧毒物质,易挥发,操作时要在毒品柜中谨慎使用。②锇酸比较昂贵,需特别节约使用。
透射电镜粉末样品制备方法
一、样品要求 1.粉末样品基本要求 (1)单颗粉末尺寸最好小于1μm; (2)无磁性; (3)以无机成分为主,否则会造成电镜严重的污染,高压跳掉,甚至击坏高压枪; 2.块状样品基本要求 (1)需要电解减薄或离子减薄,获得几十纳米的薄区才能观察; (2)如晶粒尺寸小于1μm,也可用破碎等机械方法制成粉末来观察; (3)无磁性; (4)块状样品制备复杂、耗时长、工序多、需要由经验的老师指导或制备;样品的制备好坏直接影响到后面电镜的观察和分析。所以块状样品制备之前,最好与TEM的老师进行沟通和请教,或交由老师制备。 二、送样品前的准备工作 1.目的要明确:(1)做什么内容(如确定纳米棒的生长方向,特定观察分析某个晶面的缺陷,相结构分析,主相与第二相的取向关系,界面晶格匹配等等);(2)希望能解决什么问题; 2.样品通过X-Ray粉末衍射(XRD)测试、并确定结构后,再决定是否做HRTEM;这样即可节省时间,又能在XRD 的基础上获得更多的微观结构信息。 3.做HRTEM前,请带上XRD数据及其他实验结果,与HRTEM老师进行必要的沟通,以判断能否达到目的;同时HRTEM老师还会根据您的其他实验数据,向您提供好的建议,这样不但能满足您的要求,甚至使测试内容做得更深,提高论文的档次。 三、粉末样品的制备 1.选择高质量的微栅网(直径3mm),这是关系到能否拍摄出高质量高分辨电镜照片的第一步;(注:高质量的微栅网目前本实验室还不能制备,是外购的,价格20元/只;普通碳膜铜网免费提供使用。) 2.用镊子小心取出微栅网,将膜面朝上(在灯光下观察显示有光泽的面,即膜面),轻轻平放在白色滤纸上; 3.取适量的粉末和乙醇分别加入小烧杯,进行超声振荡10~30min,过3~5 min 后,用玻璃毛细管吸取粉末和乙醇的均匀混合液,然后滴2~3滴该混合液体到微
电镜样品制备方法中英文(常用_
No.1 扫描电镜样品制备方法 样品在2.5%的戊二醛溶液中4℃固定过夜,然后按下列步骤处理样品:?倒掉固定液,用0.1M,pH7.0的磷酸缓冲液漂洗样品三次,每次15min; ?用1%的锇酸溶液固定样品1-2h; ?倒掉固定液,用0.1M,pH7.0的磷酸缓冲液漂洗样品三次,每次15min; ?用梯度浓度(包括30%,50%,70%,80%,90%和95%五种浓度)的乙 醇溶液对样品进行脱水处理,每种浓度处理15min,再用100%的乙醇处理两次,每次20 min。 ?用乙醇与醋酸异戊酯的混合液(V/V=1/1)处理样品30min,再用纯醋酸 异戊酯处理样品1-2h。 ?临界点干燥。 ?镀膜,观察。 处理好的样品在Hitachi TM-1000型扫描电镜中观察。 1.Double fixation: The specimen was first fixed with 2.5% glutaraldehyde in phosphate buffer (pH7.0) for more than 4hours; washed three times in the phosphate buffer; then postfixed with 1% OsO4 in phosphate buffer (pH7.0) for 1hour and washed three times in the phosphate buffer. 2.Dehydration: The specimen was first dehydrated by a graded series of ethanol (30%,50%, 70%, 80%, 90%, 95% and 100%) for about 15 to 20 minutes at each step, transferred to the mixture of alcohol and iso-amyl acetate (v:v=1:1) for about 30 minutes, then transferred to pure iso-amyl acetate for about 1hour. In the end, the specimen was dehydrated in Hitachi Model HCP-2 critical point dryer with liquid CO2. 3.Coating and observation: The dehydrated specimen was coated with gold-palladium and observed in Philips Model TM-1000 SEM. No.2 Negative staining of bacterium The bacterium suspension was stained by 1 to 2%solution of phosphotungstic acid (PTA) in a pH range of 6.5 to 7.0 for 15 to 30 seconds. Then, the bacterium was observed in TEM of Model JEM1230. No.3透射电镜样品制备方法 样品在2.5%的戊二醛溶液中4℃固定过夜,然后按下列步骤处理样品:?倒掉固定液,用0.1M,pH7.0的磷酸缓冲液漂洗样品三次,每次15min; ?用1%的锇酸溶液固定样品1-2h; ?倒掉固定液,用0.1M,pH7.0的磷酸缓冲液漂洗样品三次,每次15min; ?用梯度浓度(包括30%,50%,70%,80%,90%和95%五种浓度)的乙 醇溶液对样品进行脱水处理,每种浓度处理15min,再用100%的乙醇
透射电镜样品的制备方法(精)
试验材料为经过热处理后的钢材! 一、样品要求 1.粉末样品基本要求 (1)单颗粉末尺寸最好小于1μm; (2)无磁性; (3)以无机成分为主,否则会造成电镜严重的污染,高压跳掉,甚至击坏高压枪; 2.块状样品基本要求 (1)需要电解减薄或离子减薄,获得几十纳米的薄区才能观察; (2)如晶粒尺寸小于1μm,也可用破碎等机械方法制成粉末来观察; (3)无磁性; (4)块状样品制备复杂、耗时长、工序多、需要由经验的老师指导或制备;样品的制备好坏直接影响到后面电镜的观察和分析。所以块状样品制备之前,最好与TEM的老师进行沟通和请教,或交由老师制备。 二、送样品前的准备工作 1.目的要明确:(1)做什么内容(如确定纳米棒的生长方向,特定观察分析某个晶面的缺陷,相结构分析,主相与第二相的取向关系,界面晶格匹配等等);(2)希望能解决什么问题; 2.样品通过X-Ray粉末衍射(XRD)测试、并确定结构后,再决定是否做HRTEM;这样即可节省时间,又能在XRD的基础上获得更多的微观结构信息。 3.做HRTEM前,请带上XRD数据及其他实验结果,与HRTEM老师进行必要的沟通,以判断能否达到目的;同时HRTEM老师还会根据您的其他实验数据,向您提供好的建议,这样不但能满足您的要求,甚至使测试内容做得更深,提高论文的档次。 三、粉末样品的制备 1.选择高质量的微栅网(直径3mm),这是关系到能否拍摄出高质量高分辨电镜照片的第一步;(注:高质量的微栅网目前本实验室还不能制备,是外购的,价格20元/只;普通碳膜铜网免费提供使用。) 2.用镊子小心取出微栅网,将膜面朝上(在灯光下观察显示有光泽的面,即膜面),轻轻平放在白色滤纸上; 3.取适量的粉末和乙醇分别加入小烧杯,进行超声振荡10~30min,过3~5 min 后,用玻璃毛细管吸取粉末和乙醇的均匀混合液,然后滴2~3滴该混合液体到微栅网上(如粉末是黑色,则当微栅网周围的白色滤纸表面变得微黑,此时便适中。滴得太多,则粉末分散不开,不利于观察,同时粉末掉入电镜的几率大增,严重影响电镜的使用寿命;滴得太少,则对电镜观察不利,难以找到实验所要求粉末颗粒。建议由老师制备或在老师指导下制备。)
电镜样品制作(完全版)
相关仪器的信息: 透射电镜:JEOL(公司) JEM-1230(型号)TEM,产地:日本 超薄切片机:Reichert-Jung Ultracut E ultramicrotome,产地:奥地利 扫描电镜:Philips XL30 ESEM,产地:捷克 临界点干燥仪:Hitachi HCP-2 Critical point dryer,产地:日本 真空喷镀仪:Eiko IB5 ion coater,产地:日本 包埋剂:SPI-CHEM Spurr resin,产地:USA TEM样品包埋块制作有关注意事项 一、取材: 1、动作迅速:组织离体后,应将其快速放入固定液中,使组织 细胞尽可能保持原来的生活状态; 2、减少损伤:选择锋利切割器械,减少牵拉或挤压组织; 3、组织块大小:一般要小于1mm3。 二、固定:固定是指用化学固定剂或一些物理方法迅速杀死细胞 的过程,目的是尽可能保持细胞的原有生活状态,不发生位移,减少组织结构变化。固定剂的主要作用是使蛋白质、脂质等生物大分子发生某种交联。 1、用固定液固定的样品若漂浮在溶液表面,应通过抽真空的方 法让样品沉入溶液中 2、使用锇酸的注意事项 I 通风橱中的锇酸溶液为2%的储备液,使用之前需用0.2MPBS或
二甲砷酸盐缓冲液稀释成1%的锇酸溶液。 II 锇酸为剧毒、极易挥发的试剂,必须在通风橱中操作,废液必须收集在密闭容器中。第一次使用锇酸的同学,请务必获得老师或其它熟悉操作人员的指导。 三、漂洗:应彻底漂洗干净,减少固定液与固定液或与脱水剂之间的 反应。 四、脱水:脱水是将组织中的游离水彻底清除的过程。由于常用的包 埋剂大都是非水溶性树脂,只有将生物组织中的游离水清除干净,包埋剂才能浸入组织。 脱水过程中应注意: I 逐级脱水而不能急剧脱水; II更换溶液时动作要快,特别是不要让组织离开溶液,否则会在组织内外产生气泡; III脱水过程中若要长时间停留或过夜,应放在70%脱水剂中,并在4℃保存。 五、渗透:渗透就是用包埋剂逐渐取代组织中的脱水剂,使细胞内外 空隙被包埋剂所填充。包埋剂通常由树脂、硬化剂、增塑剂及催化剂4种试剂按一定比例配制而成。 包埋剂配制及使用过程中的注意事项: I 所有容器及玻璃棒等应是清洁和干燥的; II配制过程中应搅拌均匀,使用过程中应避免异物,特别是水、乙醇、丙酮等混入包埋剂;
电子显微镜生物样品的制备实验
电子显微镜生物样品的制备实验 一、目的要求 学习并掌握制备微生物及核酸电镜样品的基本方法。 二、电子显微镜与光学显微镜的主要区别 显微镜的分辨率取决于所用光的波长,1933年开始出现的电子显微镜正是由于使用了波长比可见光短得多的电子束作为光源,使其所能达到的分辨率较光学显微镜大大提高。而光源的不同,也决定了电子显微镜与光学显微镜的一系列差异(表Ⅲ-1)。表Ⅲ-1 根据电子束作用于样品的方式的不同及成像原理的差异,现代电子显微镜已发展形成了许多种类型,目前最常用的是透射电子显微镜(transmission electron microscope)和扫描电子显微镜(scanning electron microscope),前者总放大倍数可在1000~1000000倍范围内变化,后者总放大倍数可在20~300000倍之间变化。本实验主要介绍这两种显微镜样品的制备。 三、器材 1.菌种大肠杆菌(大肠埃希氏菌,Escherichia coli)斜面。 2.溶液或试剂醋酸戊脂,浓硫酸,无水乙醇,无菌水,2%磷钨酸钠(pH6.5~8.0)水溶液,0.3%聚乙烯甲醛(溶于三氯甲烷)溶液,细胞色素c,醋酸铵,质粒pBR322。 3.仪器或其他用具普通光学显微镜,铜网,瓷漏斗,烧杯,平皿,无菌滴管,无菌镊子,大头针,载玻片,细菌计数板,真空镀膜机,临界点干燥仪等。 四、操作步骤 (一)透射电镜的样品制备及观察 1.金属网的处理 光学显微镜的样品是放置在载玻片上进行观察。而在透射电镜中,由于电子不能穿透玻璃,只能采用网状材料作为载物,通常称为载网。载网因材料及形状的不同可分为多种不同的规格,其中最常用的是200~400目(孔数)的铜网。网在使用前要处理,除去其上的污物,否则会影响支持膜的质量及标本照片的清晰度。本实验选用的是400目的铜网,可用如下方法进行处理:首先用醋酸戊酯浸漂几小时,再用蒸馏水冲洗数次,然后再将铜网浸漂在无水乙醇中进行脱水。如果铜网经以上方法处理仍不干净时,可用稀释的浓硫酸(1:1)浸1~2分钟,或在1% NaOH 溶液中煮沸数分钟,用蒸馏水冲洗数次后,放入无水乙醇中脱水,待用。 2.支持膜的制备 在进行样品观察时,在载网上还应覆盖一层无结构、均匀的薄膜,否则细小的样品会从载网的孔中漏出去,这层薄膜通常称为支持膜或载膜。支持膜应对电子透明,其厚度一般应低于20nm;在电子束的冲击下,该膜还应有一定的机械强度,能保持结构的稳定,并拥有良好的导热性;此外,支持网在电镜下应无可见的结构,且不与承载的样品发生化学反应,不干扰对样品的观察,其厚度一般为15nm左右。支持膜可用塑料膜(如火棉膜、聚乙烯甲醛膜等),也可以用碳膜或者金属膜(如铍膜等)。常规工作条件下,用塑料膜就可以达到要求,而塑料膜中火棉胶膜的制备相对容易,但强度不如聚乙烯甲醛膜。 (1)火棉胶棉的制备在一干净容器(烧杯、平皿或下带止水夹的瓷漏斗)中放入一定量的无菌水,用无菌滴管吸2%火棉胶醋酸戊酯溶液,滴一滴于水面中央,勿振动,待醋酸戊
透射电镜样品制备方法
?透射电子显微镜成像时,电子束是透过样品成像。 ?由于电子束的穿透能力比较低,用于透射电子显微镜分析的样品必须很薄。 ?根据样品的原子序数大小不同,一般在50~500nm之间。 透射电镜样品的要求: ?1. 样品必须对电子束透明。 ?2. 所制得样品必须具有代表性,以真实反映所分析材料的特征。 主要方法: 粉末样品、复型、离子减薄、电解双喷。
?透射电镜观察用的样品很薄,需放在专用的样品铜网上。 ?透射电子显微镜使用的铜网一般直径为3毫米,上面铳有许多微米大小的孔,在铜网上覆盖了一层很薄的火棉胶膜并在上面蒸镀了碳层以增加其膜的强度,被分析样品就承载在这种支撑膜上。
样品铜网的作用: ?承载样品,并使之在物镜极靴孔内平移、倾斜、旋转,寻找观察区。 ?样品通常放在外径3mm ,200目方孔或圆孔的铜网上,铜网牢固夹持在样品座中保持好的热、点接触,减少因电子照射引起的热或电荷积累而产生样品漂移或损伤。 样品台
透射电子显微镜样品制备 电镜观察时样品受到的影响: (1)真空的影响。含有挥发溶剂或易升华的试样必须冷冻后观 察。 (2)电子损伤的影响。试样在电镜中受到l0-3~1A/cm2的电子 束照射,电子束的能量部分转化为热,使试样内部结构或外形发生变化或污染。观察有机物或聚合物试样时,为防止电子束对试样的损伤和污染,应提高电压。 (3)电子束透射能力的影响。由于电子束透射能力较弱,一般 100kv加速电压时,试样厚度必须在20~200nm之间。
粉末样品制备 ?随着材料科学的发展,超细粉体及纳米材料发展很快,而粉末的颗粒尺寸大小、尺寸分布及形态对最终制成材料的性能有显著影响,因此,如何用透射电镜来观察超细粉末的尺寸和形态便成了电子显微分析的一的一项重要内容。 ?其关键工作是是粉末样品的制备,样品制备的关键是如何将超细粉的颗粒分散开来,使其均匀分散到支持膜上,各自独立而不团聚。
粉末样品 透射电镜试样制备
透射电镜试样制备 一、样品要求 1.粉末样品基本要求 (1)单颗粉末尺寸最好小于1μm; (2)无磁性; (3)以无机成分为主,否则会造成电镜严重的污染,高压跳掉,甚至击坏高压枪; 2.块状样品基本要求 (1)需要电解减薄或离子减薄,获得几十纳米的薄区才能观察; (2)如晶粒尺寸小于1μm,也可用破碎等机械方法制成粉末来观察; (3)无磁性; (4)块状样品制备复杂、耗时长、工序多、需要由经验的老师指导或制备;样品的制备好坏直接影响到后面电镜的观察和分析。所以块状样品制备之前,最好与TEM的老师进行沟通和请教,或交由老师制备。 二、送样品前的准备工作 1.目的要明确:(1)做什么内容(如确定纳米棒的生长方向,特定观察分析某个晶面的缺陷,相结构分析,主相与第二相的取向关系,界面晶格匹配等等);(2)希望能解决什么问题; 2.样品通过X-Ray粉末衍射(XRD)测试、并确定结构后,再决定是否做HRTEM;这样即可节省时间,又能在XRD的基础上获得更多的微观结构信息。 3.做HRTEM前,请带上XRD数据及其他实验结果,与HRTEM老师进行必要的沟通,以判断能否达到目的;同时HRTEM老师还会根据您的其他实验数据,向您提供好的建议,这样不但能满足您的要求,甚至使测试内容做得更深,提高论文的档次。 三、粉末样品的制备 1.选择高质量的微栅网(直径3mm),这是关系到能否拍摄出高质量高分辨电镜照片的第一步;(注:高质量的微栅网目前本实验室还不能制备,是外购的,价格20元/只;普通碳膜铜网免费提供使用。) 2.用镊子小心取出微栅网,将膜面朝上(在灯光下观察显示有光泽的面,即膜面),轻轻平放在白色滤纸上; 3.取适量的粉末和乙醇分别加入小烧杯,进行超声振荡10~30min,过3~5 min后,用玻璃毛细管吸取粉末和乙醇的均匀混合液,然后滴2~3滴该混合液体到微栅网上(如粉末是黑色,则当微栅网周围的白色滤纸表面变得微黑,此时便适中。滴得太多,则粉末分散不开,不利于观察,同时粉末掉入电镜的几率大增,严重影响电镜的使用寿命;滴得太少,则对电镜观察不利,难以找到实验所要求粉末颗粒。建议由老师制备或在老师指导下制备。) 4.等15 min以上,以便乙醇尽量挥发完毕;否则将样品装上样品台插入电镜,将影响电镜的真空。 四、块状样品制备 1.电解减薄方法 用于金属和合金试样的制备。(1)块状样切成约0.3mm厚的均匀薄片;(2)用金刚砂纸机械
电镜样品制备方法中英文(常用
电镜样品制备方法中英文(常用 倒掉固定液,用0、IM, pII7、0的磷酸缓冲液漂洗样品三次,每次15min;用1%的餓酸溶液固定样品l-2h;倒掉固定液,用0、IM, pII7、0的磷酸缓冲液漂洗样品三次,每次15min;用梯度浓度(包括30%,50%, 70%, 80%, 90%和95%五种浓度)的乙醇溶液对样品进行脱水处理,每种浓度处理15min,再用100% 的乙醇处理两次,每次20 mine 用乙醇与醋酸异戊酯的混合液(V/V二1/1)处理样品30min, 再用纯醋酸异戊酯处理样品l-2h o 临界点干燥。 镀膜,观察。处理好的样品在Hitachi TM-1000型扫描电镜 中观察。1 Double fixation: The specimen was first fixed with2、 5% glutaraldehyde in phosphate buffer (pII7、 0)for more than4hours; washed three times in the phosphate buffer; then postfixed withl% 0s04 in phosphate buffer (pII7、0) forlhour and washed three times in the phosphate buffer、 2、 Dehydration: The specimen was first dehydrated by a graded series of ethanol (30%,50%,70%,80%,90%,95% and100%)
for about 15 to20 minutes at each step, transferred to the mixture of alcohol and iso-amyl acetate (v:v=l:1) for about30 minutes, then transferred to pure isoamyl acetate for aboutlhourIn the end, the specimen was dehydrated in Ilitachi Model HCP-2 critical point dryer with liquid C02、 3、 Coating and observation: The dehydrated specimen was coated with gold-palladium and observed in Philips Model TM-1000 SEM、 No、 2 Negative staining of bacteriumThe bacterium suspension was stained byl to2%solution of phosphotungstic acid (PTA) in a pH range of6、 5 to7、 0 forl5 to30 seconds. Then, the bacterium was observed in TEM of Model JEM12 30、No、3透射电镜样品制备方法样品在2、5%的戊二醛溶液 中4工固定过夜,然后按下列步骤处理样品: 倒掉固定液,用0、IM, pII7、0的磷酸缓冲液漂洗样品三 次,每次15min;用1%的餓酸溶液固定样品l-2h;倒掉固定液,用0、IM, pH7、0的磷酸缓冲液漂洗样品三次,每次15min;用梯度 浓度(包括30%,50%, 70%, 80%, 90%和95%五种浓度)的乙醇溶液对样品进行脱水处理,每种浓度处理15min,再用100% 的乙醇处理 一次,每次20min;最后过度到纯丙酮处理20min o 用包埋剂与丙酮 的混合液(V/V二1/1)处理样品lh;用包埋剂与丙酮的混合液 (V/V=3/l)处理样品3h;纯包埋剂处理样品过夜;将经过渗透处
【材料课堂】TEM透射电镜的样品制备方法
【材料课堂】TEM透射电镜的样品制备方法 材料科学与工程点击上方「材料科学与工程」快速关注材料类综合、全面、专业的微信平台 1TEM 样品台样品台的顶端2对样品的要求 1. 样品一般应为厚度小于100nm的固体。 2. 感兴趣的区域与其它区域有反差。 3. 样品在高真空中能保持稳定。 4. 不含有水分或其它易挥发物,含有水分或其他易挥发物的试样应先烘干除去。 5. 对磁性试样要预先去磁,以免观察时电子束受到磁场的影响。 TEM样品常放置在直径为3mm的200目样品网上,在样品网上常预先制作约20nm厚的支持膜。 3纳米粉末样品的制备方法 1. 纳米颗粒都小于铜网的小孔,因此要先制备对电子束透明的支持膜。 2. 将支持膜放在铜网上,再把粉末放在膜上,送入电镜分析。 3. 粉末或颗粒样品制备的关键取决于能否使其均匀分散到支持膜上。 4. 用超声波分散器将需要观察的粉末在分散介质(不与粉末发生作用)中分散成悬浮液。 5. 用滴管滴几滴在覆盖有支持膜的电镜铜网上,待其干燥(或用滤纸吸干)后, 即成为电镜观察用的粉末样品。 6. 微米粉末样品通过研磨转为纳米颗粒,如催化剂等。4块状样品的制备方法4.1超薄切片法
超薄切片方法多用于生物组织、高分子和无机粉体材料等。超薄切片过程图4.2离子轰击减薄法 离子轰击减薄法多用于矿物、陶瓷、半导体及多相合金等。 1. 将待观察的试样按预定取向切割成薄片,再经机械减薄抛光等过程预减薄至30-40μm的薄膜。 2. 把薄膜钻取或切取成尺寸为2.5-3mm的小片。 3. 装入离子轰击减薄装置进行离子轰击减薄和离子抛光。 原理: 在高真空中,两个相对的冷阴极离子枪,提供高能量的氩离子流,以一定角度对旋转的样品的两面进行轰击。 当轰击能量大于样品材料表层原子的结合能时,样品表层原子受到氩离子击发而溅射、经较长时间的连续轰击、溅射,最终样品中心部分穿孔。 穿孔后的样品在孔的边缘处极薄,对电子束是透明的,就成为薄膜样品。 4.3电解抛光减薄法 电解抛光减薄方法适用于金属与部分合金。4.4聚焦离子束法 适用于半导体器件的线路修复和精确切割。 聚焦离子束系统(FIB),利用源自液态金属镓的离子束来制备样品。 通过调整束流强度,FIB可以对样品的指定区域进行快速和
电镜样品制备
电子显微镜样品的制备 一、透射电镜样品超薄切片常规制作规程 1.取材:根据实验目的取材,要求部位准确,体积小于2mm3 2.醛类固定:用2%-3%的戊二醛固定2小时; 3.清洗:用磷酸缓冲液清洗3次,每次10min; 4.锇酸固定:用1%-2%的锇酸固定2~3小时; 5.清洗:用磷酸缓冲液清洗3次,每次10 min; 6.脱水:用50%、70%、80%、90%乙醇梯度脱水各15min,再用100%乙醇脱水3次,每次30min; 7.置换:用环氧丙烷或丙酮置换3次,每次30min; 8.浸渍:10hr以上,浸渍过程如下: ①丙酮:包埋剂=3:1的浸渍液浸渍(动物样品1hr,植物样品2~3hr); ②丙酮:包埋剂=1:1的浸渍液浸渍(动物样品1hr,植物样品2~3hr); ③丙酮:包埋剂=1:3的浸渍液浸渍(动物样品4hr,植物样品12~24hr); ④纯包埋剂浸渍(动物样品4hr,植物样品12~24hr); 9.包埋:将样品放入盛有纯包埋剂的包埋板中; 包埋剂配方中Epon812,MNA,DDSA,DMP-30的比例见附表 10.聚合:将包埋板置于40C、60℃条件下各聚合48hr; 11.修块:将包埋头修成梯形,且样品表面积小于0.2mm×0.2mm 12.超薄切片:切片厚度50-90nm 13.染色:铀染色 5~15min 清洗 铅染色 5~10min 清洗 二、扫描电镜样品常规制备规程 1.取材:根据实验目的取材,要求部位准确 2.清洗:用磷酸缓冲液反复清洗样品表面的灰尘、杂质等附着物 3.固定:用2%-3%的戊二醛固定2小时 4.清洗:用磷酸缓冲液清洗3次,每次10 min 5.脱水:用50%、70%、80%、90%乙醇梯度脱水各15min,再用100%乙醇脱水3次,每次30min; 6.置换:用叔丁醇置换3次,每次30min 7.干燥:用冷冻干燥仪干燥样品 8.粘样:用双面胶带将样品粘到样品台上 9.镀膜:用离子溅射仪给样品镀10nm金膜
透射电镜超薄切片步骤
1 玻璃刀(glass knife) 與刀口水槽(trough) 製作 玻璃刀需於切片當天製作,請於切片當天預留0.5-1 h。 方法步驟: 1)首先取一乾淨的玻璃條,粗造面朝下(由玻璃條側面觀之)。利用製刀機將玻璃條(glass strip) 切割成一英吋大小的 正方形,再由對角線(約略偏斜) 切割一裂痕,自兩側及底部加壓使玻璃斷裂為二即成。理想的刀口應呈現平整均勻,在解剖顯微鏡下無鋸齒狀缺刻(圖1.5.1)。 施加壓力於經鑽石刀劃過的玻璃條時,需慢慢的添加壓力。當切口出現一半月型的陰影或白影(視觀察角度而定) 時,即停止繼續增加壓力,使切口自行持續擴大至斷裂。經此過程做出的玻璃刀有較佳的品質。 圖1.5.1 玻璃刀的製作 (A) LKB700製刀機。(B) 利用製刀機上的鑽石刀在玻璃條上略做切割。(C) 旋轉製刀機右側旋鈕,自玻璃條 兩側及下方均勻施加壓力,使其斷裂。(D) 及(E) 每一正方形玻璃條可製作2把玻璃刀,每把刀各自有刀鋒及 刀座。第一把刀的刀鋒恰好緊鄰第二把刀的刀座。(F) 製作完成的玻璃刀可以在顯微鏡下檢查刀鋒部分,理想 的刀鋒應為平滑無缺刻。整把刀最適合切片的部分為Z區,約佔1/3;S區不用於切片;而E區的使用需視刀 鋒的品質而定。(圖片A-C, E取自生物電子顯微鏡學, 圖片D, F取自Methods of Preparation for Electron Microscopy, An Introduction for the Biomedical Sciences) 2)將銀膠帶的一端貼於刀口下方並與底部平行後,另一端圍繞成一平滑的弧形貼於另一側,圈出一船型水槽。過程中勿 用手接觸用以形成水槽,帶有黏性的膠帶面,以免將手上的油脂黏於其上(圖1.5.2 A-B)。 3)以刀片沿玻璃將多餘的膠帶切除,然後於水槽下緣與玻璃刀接觸面以蠟或指甲油封合,避免水滲漏(圖1.5.2 C),待 乾燥後即可使用。 圖1.5.2 刀口水槽的製(圖片取自生物電子顯微鏡學) 2、樣品塊修整(trimming) 包埋完成後的樣品塊須先經過修整,將樣品塊尖端修整成適當的大小及形狀,並將要觀察的部份露出。習慣上將樣品塊修整成平頂的金字塔型(從側面觀察),頂部與底部略成梯形(從上方觀察),大小應小於0.5 mm,金字塔高度不宜超過0.2 mm。 將樣品塊修整成平頂的金字塔型,是為了讓樣品在切片過程中得到最佳的支持力。高度過大,切片時容易發生震動,在切片上留下深淺交替的平行條紋,或導致樣品塊前端斷裂。 較小的樣品塊切面較容易進行超薄切片,但過小的切片在電子顯微鏡下可觀察到的東西較少。 方法步驟: 1) 將樣品塊放置在解剖顯微鏡下,以刀片(two-edged razor blade) 水平的削去尖端直到組織露出(圖1.4.1, 步驟1)。 2) 修出梯形的上下底。先將刀片以垂直方向切下,再將刀鋒以水平朝向自己的方式緩慢前進,至前一刀垂直切下處即可除去不要的部份(圖1.4.1, 步驟2-3)。注意力量的控制。 3) 修出梯形的兩邊,方式同上(圖1.4.1, 步驟4-5)。