蛋白纯化操作

His-tag融合蛋白的纯化
The following protocols is for His Bind resin
His Bind Purification
1) Express and harvest cells. Resuspend cells in Binding Buffer. Lyse cells.
2) Prepare resin (~20- 25 ml?) in column - wet column and frit, transfer resin slurry to column. Wash with water (3 volume), Charge Buffer (5 volumn), Binding Buffer (3 volume).
3) Load column with cell lysate, at a rate of ~10 column volumn per hour.
4) Wash column (10 X volumn) with Binding Buffer
5) Wash column (6 X volumn) with Wash Buffer
6) Elute protein (6 X volume) with Elute Buffer (strip buffer can also be used to elute). Alternatively elute first with lower concentration of imidazole at 0.25 M.
7) To store column, wash with Strip Buffer (3 X volumn) and stored in Strip Buffer at 4 °C.
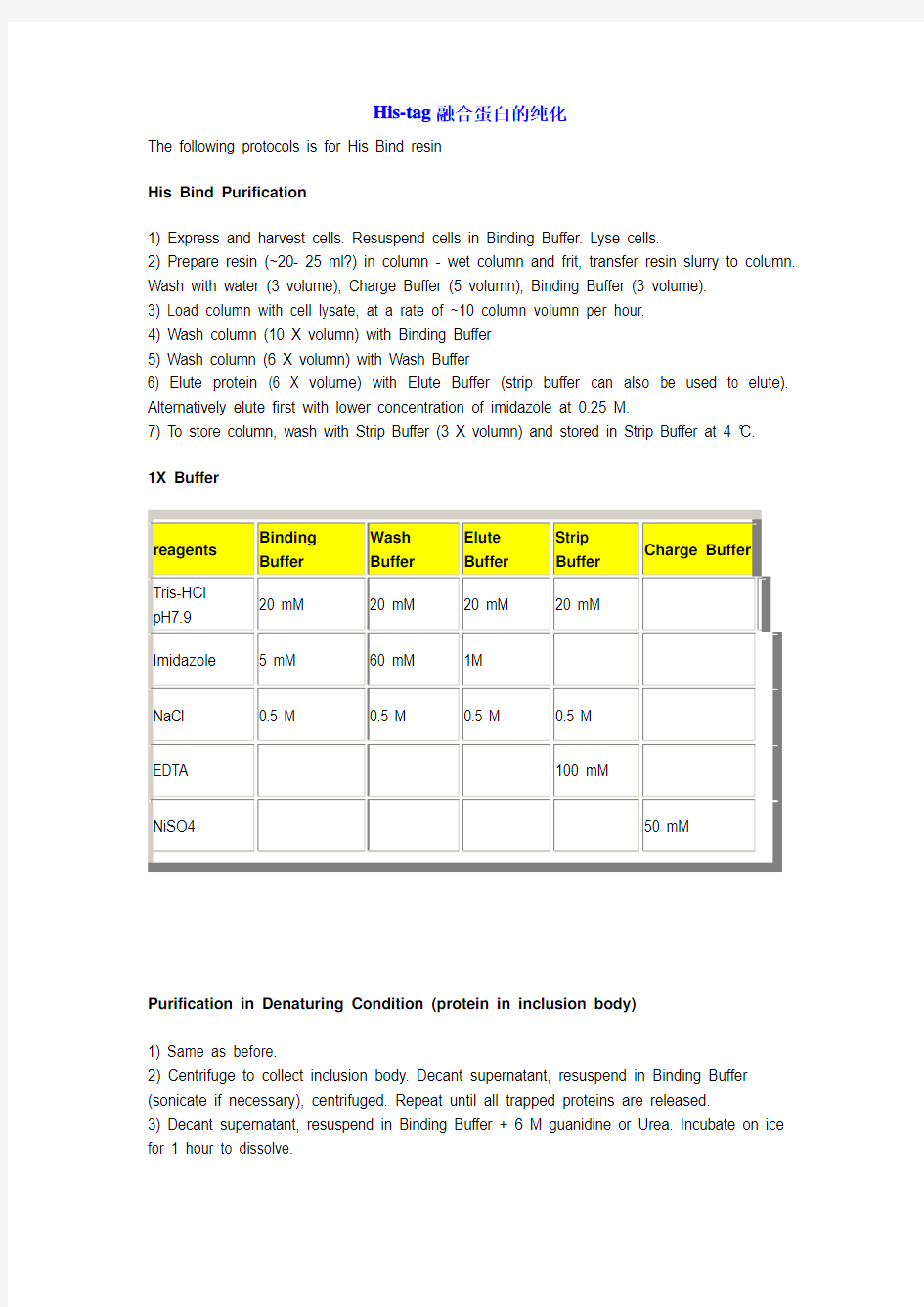
1X Buffer
Purification in Denaturing Condition (protein in inclusion body)
1) Same as before.
2) Centrifuge to collect inclusion body. Decant supernatant, resuspend in Binding Buffer (sonicate if necessary), centrifuged. Repeat until all trapped proteins are released.
3) Decant supernatant, resuspend in Binding Buffer + 6 M guanidine or Urea. Incubate on ice for 1 hour to dissolve.
4) Centrifuged at 39,000 g for 20 min. Filter supernatant (0.45 micron membrane).
5) Load onto column. Purification same as before, except that all buffer contain denaturant, and lower imidazole concentration in Wash Buffer (20 mM) and Elute Buffer (~300mM) (dilute Wash and Elute Buffer with Binding Buffer). Alternatively, you can wash and elute the protein without imidazole at low pH (wash at pH 6.5, elute at pH5.9 or pH4.5 if the protein failed to elute at the hight pH)
Regeneration of Column
- when flow rate is slow and column resin doesn't turn blue-green when charged
1) Wash with 2 vol 6 M guanidine-HCl, then 3 vol water.
2) Wash with 1 vol 2% SDS.
3) Wash with 1 vol each of 25%, 50% and 75% ethanol, then 5 vol of 100% ethanol, followed by 1 vol each of 75%, 50% and 25% ethanol.
4) Wash with 1 vol water, then 5 volume 100 mM EDTA (pH 8.0).
5) Wash with 3 vol water, then 3 vol 20% ethanol.
6) Store at 4 °C.
Note:
1) Do not use ?ME, DTT or EDTA in buffer.
2) Higher amount of imidazole may be used in the wash buffer (100 mM) but some protein may be eluted.
3) You can also elute the protein with salt concentration gradient or using lower pH.
4) It's sometimes useful to introduce a couple of glycines between your protein and the His-tag which allow the his-tag to be fully exposed for effective binding to resin (if your vector doesn't have a short linker sequence between the HIs-tag and your protein that is).
His-tag融合蛋白纯化中Ni柱的使用Materials and reagents
Extraction buffer
Preparation of the magnetic beads
Procedure
GST融合蛋白纯化方法
Abstract: Many people have vented out frustration over insoluble GST-fused proteins. This is a protocol for enzymatically active soluble GST-fused proteins. All GST-fused proteins are rendered soluble with this technique though enzyme activitiy can range from 30-90%.
Materials and Reagents
1.STE Buffer
10 mM Tris-HCl, pH 8.0
1 mM EDTA
150 mM NaCl
2.Lysozyme solution
10 mg/ml in water (make fresh)
3.PBS
4.Elution Buffer
50 mM Tris.Cl, pH 9.0
20 mM GSH
5.10% Sarkosyl in STE Buffer
6.10% Triton X-100 in STE Buffer
7. 1 M DTT
8.100 mM IPTG
Procedure
Day 1
1.Set up an overnight culture in 50 ml 2XTY with 150 mg/ml of ampicillin.
Day 2
1.Seed 5 ml of overnight culture to 500 ml 2XTY with 150 mg/ml of ampicillin.
2.Grow at 37o C to an A600 of 0.6 to 0.8.
3.Induce with 0.1 mM to 2 mM of IPTG. Grow for 3 hr at 37o C or grow overnight at room
temperature.
Lower IPTG concentrations and lower growing temperatures tend to produce greater solubility at the expense of yield.
4.Pellet cells by centrifuging at 3000 g, 4o C for 10 min. Decant media and resuspend cells
in 30 ml ice-cold PBS to wash. Transfer to a 40-ml Oak Ridge tube and centrifuge at 3000 g, 4o C for 10 min. Decant PBS.
5.This is a convenient point to stop and to store pellets at -80o C. Else continue to lyse
cells.
6.Thaw pellet on ice if cells are frozen else proceed to the next step.
7.Resuspend pellet in 10 ml of ice cold STE Buffer.
8.Add 100 ml of freshly prepared lyozyme solution, incubate on ice for 15 min. Just
before sonication, add 100 ml of 1 M DTT and 1.4 ml of 10% Sarkosyl. Mix thoroughly by inversion and sonicate for a total time of 1 min.
9.Centrifuge 16,000 rpm for 20 min on the SS34 rotor to pellet debris. Transfer
supernatant to a 50-ml conical tube and discard the pellet. Add 4 ml of 10% Triton X-100 and top up with STE Buffer to 20 ml. The effective concentration of Sarkosyl and Triton X-100 will be 0.7% and 2% respectively. Incubate at room temperature for
30 min.
10.Pour the lysate to 1 ml bed of prepared Glutathione Sepharose in PBS. Incubate at
room temperature for 30 min to 1 hr with agitation.
To prepare the 50% slurry, shake up the media and pipette 2 ml to a 50 ml
tube. Fill to 50 ml with PBS, invert tube a few times. Centrifuge to 2000 rpm
on a swing bucket centrifuge then switch off. Carefully suck off PBS and
resuspend beads with 1 ml of PBS.
11.Wash the beads with 3 X 50 ml of PBS. Finally resuspend in 5 ml of PBS. Pour to a
dispo-column. Wash the 50-ml conical tube with an additional 5 ml of PBS. Pool with
the first 5 ml in the dispo-column.
To wash, use the same centrifugation technique for preparing the beads. When transferring beads to column, do not pipette but pour. The beads tend to stick
to pipette tips.
12.If desired, elute with 10 x 1 ml fractions of Elution Buffer. Determine desired fractions
with SDS PAGE.
Reference
Frangioni and Neel.Anal. Biochem. 210, 179-187 (1993)
酵母中GST融合蛋白纯化方法
? 5 ml overnight culture of your favorite yeast in your favorite medium.
?Inoculate 50 ml and grow 30o C shaking O/N until OD600 = 0.8 to 1.2. For SCD cultures use 1/500 and 1/1500 dilutions.
?Optional: add alpha-factor to 2.5 μM. Continue shaking at 30o C for 60 min.
?Add 1 M NaN 3 to 10 mM (final concentration) and move cultures to ice. Everything must remain cold from here on out.
?Spin cells 3 K, 10 min at 4o C.
?Discard supe and resuspend cells with 0.5 ml 10 mM NaN3. Transfer to 1.5 ml microfuge tube (not autoclaved).
?Spin 3 K, 10 min at 4o C. Alternatively spin 8 K, 1 min at room temperature.
?Discard supe. Optional: freeze pellet at -80o C.
?Resuspend in 1 ml of cold 10 mM NaN3.
?Measure OD600. Adjust volumes so that there are an equal number of cells in each sample. A total OD600 of 30 per sample is best.
?Wash with 1 ml of Lysis Buffer.
?Spin 3 K 10 min at 4o C and discard supe.
?Resuspend in 400 ul Lysis Buffer.
?Add a scoop of glass beads to a 0.5 ml PCR tube. Transfer cell lysate to the PCR tube
?Vortex 1 min, 4X. Keep samples cold between vortexing.
?Poke a hole in the bottom of the tube and spin cell lysate in a new microfuge tube 1.5 K or 500 X g, 10 min., 4o C.
?Transfer liquid from bottom tube into a new microfuge tube.
?Spin again 1.5 K, 10 min, 4o C and again transfer liquid into a new microfuge tube.
?Add Triton X-100 to 1.5 % and rock for 60 min at 4o C.
?Spin (3 K, 10 min, 4o C) and transfer the supe to a new microfuge tube.
?Remove 30 ul of liquid and add 30 ul 2X SDS PAGE Sample buffer. This will reflect protein content before Glutathione purification.
?To the remaining liquid, add 100 ul 40 % slurry of Glutathione beads and mix at 4o C for 2 h (overnight is usually fine). Glutathione beads should be prewashed 3 X with PBS and 1 X with lysis buffer before resuspending as a 40 % slurry in lysis buffer.
?Wash glutathione beads five times with PBS, 1 % Triton X-100, 300 mM NaCl at RT. Spin 2 K, 5 min, at room temperature. Rock sample for 5 min between washes. Change tubes after the first wash to reduce nonspecific binding to the tube itself.
?Resuspend in SDS-PAGE Sample buffer.
o Alternatively elute 3 times with 1-2 column volumes of 5-10 mM reduced glutathione, 50 mM Tris pH 8, and mix with 6X SDS-PAGE sample buffer
before stripping the beads with SDS-PAGE sample buffer.
?Heat to 100o C for 10 min. Then store at -20o C. Protein is ready to be run on SDS-PAGE Gel.
NOTES
Thanks to Paul DiBello and Jiyoung Cha for their refinements of this protocol.
* Indicates a correction from an eariler version of the protocol.
1. For lysis in the presense of GDP and GTP I use a final concentration of 10 uM GDP or 20 uM GTPgammaS and a final concentration of 3 mM MgCl2 in the lysis buffer.
2. You can substitute protease inhibitor cocktail (Sigma P8215) for individual protease inhibitors (AEBSF, leupeptin, pepstatin, benzamidine, aprotinin).
3. For lysis to determine phosphorylation, you may wish to add more phosphatase inhibitors (in addition to beta-glycerolphosphate and Na-o-vanadate) to the lysis buffer, or you may wish to omit phosphatase inhibitors altogether:
50mM Na-M-Vanadate 200ml 0.5mM
100mM
2 ml 10mM
Na-pyrophosphate
2mg/ml Phosvitin 10μl 1μg/ml
蛋白质的纯化方法
蛋白质纯化的方法 蛋白质的分离纯化方法很多,主要有: (一)根据蛋白质溶解度不同的分离方法 1、蛋白质的盐析 中性盐对蛋白质的溶解度有显著影响,一般在低盐浓度下随着盐浓度升高,蛋白质的溶解度增加,此称盐溶;当盐浓度继续升高时,蛋白质的溶解度不同程度下降并先后析出,这种现象称盐析,将大量盐加到蛋白质溶液中,高浓度的盐离子(如硫酸铵的SO4和NH4)有很强的水化力,可夺取蛋白质分子的水化层,使之“失水”,于是蛋白质胶粒凝结并沉淀析出。盐析时若溶液pH在蛋白质等电点则效果更好。由于各种蛋白质分子颗粒大小、亲水程度不同,故盐析所需的盐浓度也不一样,因此调节混合蛋白质溶液中的中性盐浓度可使各种蛋白质分段沉淀。 影响盐析的因素有:(1)温度:除对温度敏感的蛋白质在低温(4度)操作外,一般可在室温中进行。一般温度低蛋白质溶介度降低。但有的蛋白质(如血红蛋白、肌红蛋白、清蛋白)在较高的温度(25度)比0度时溶解度低,更容易盐析。(2)pH值:大多数蛋白质在等电点时在浓盐溶液中的溶介度最低。(3)蛋白质浓度:蛋白质浓度高时,欲分离的蛋白质常常夹杂着其他蛋白质地一起沉淀出来(共沉现象)。因此在盐析前血清要加等量生理盐水稀释,使蛋白质含量在2.5-3.0%。 蛋白质盐析常用的中性盐,主要有硫酸铵、硫酸镁、硫酸钠、氯化钠、磷酸钠等。其中应用最多的硫酸铵,它的优点是温度系数小而溶解度大(25度时饱和溶液为4.1M,即767克/升;0度时饱和溶解度为3.9M,即676克/升),在这一溶解度范围内,许多蛋白质和酶都可以盐析出来;另外硫酸铵分段盐析效果也比其他盐好,不易引起蛋白质变性。硫酸铵溶液的pH常在4.5-5.5之间,当用其他pH值进行盐析时,需用硫酸或氨水调节。 蛋白质在用盐析沉淀分离后,需要将蛋白质中的盐除去,常用的办法是透析,即把蛋白质溶液装入秀析袋内(常用的是玻璃纸),用缓冲液进行透析,并不断的更换缓冲液,因透析所需时间较长,所以最好在低温中进行。此外也可用葡萄糖凝胶G-25或G-50过柱的办法除盐,所用的时间就比较短。
AKTA蛋白纯化系统操作
AKTA蛋白纯化系统操作 AKTA蛋白纯化系统是当前蛋白纯化工作经常用到的一组设备,自动化程度很高。AKTA系统依据不同的配置,可以分为AKTA EXPLORER、AKTA PILOT、AKTA PURIFIER等多种型号的设备。以下以AKTA EXPLORER为例简单介绍AKTA蛋白纯化系统的一般操作。 1、认识AKTA。 AKTA explorer 是为方法开拓及研究应用而设计的全自动液相色谱系统。该色谱系统的分离装置有三个主要组件,在底部平台的左侧整齐堆起(Fig 1)。它们是: FIG 1、AKTA EXPLORER主机 ? Pump-900 为双通道高效梯度泵系列。在AKTAexplorer 100,流速范围0.01-100 ml/min,压力高达10 Mpa(泵名为P-901)。在AKTA explore10,流速范围0.001-10 ml/min,压力高达25 Mpa(泵名为P-903)。 ? Monitor UV-900,同时监控190-700 nm 范围内高达3 个波长的多波长紫外-可见(UV-Vis)监测器。(针对部分AKTA PURIFIER机型,尚有UPC-900监测器可供选择,光源为汞灯光源,一次可以监控一个波长,安装滤光片后,可以在选择的波长范围内进行切换。)? Monitor pH/C-900,在线电导和pH 监测的组合监测器。 Fig 2、AKTA EXPLORER硬件模式图
AKTA EXPLORER系统的主要组成部件可以用模式图表示(Fig 2)。组成部件,如混合器、柱及不同的阀安装在右边部分。打开装阀的门可全部看到。柱被挂在装阀的门的外侧。 分离装置由UNICORN 软件控制。软件安装于一独立的电脑主机之中,在电脑与色谱系统之间的通信由数据采集装置CU950进行控制。 2、一般操作 2.1 开机 按位于底部平台前左侧的ON/OFF 按钮,打开色谱系统,然后打开电脑电源。待仪器自检完毕(CU950上面的3个指示灯完全点亮并不闪烁)。双击桌面上UNICORN图标,进入操作界面。UNICORN的操作界面分为四个窗口(Fig 3) Fig 3、Unicorn的操作界面 2.2准备工作溶液和样品 所有的工作溶液和样品必须经过0.45μm的滤膜过滤,样品也可高速离心后取上清备用。当缓冲液中含有有机溶剂(如乙腈、甲醇),需在使用前用低频超声脱气10min。 2.3清洗及管道准备 首先将A泵的进液管道(A1)放入缓冲液或平衡液中,将B泵的进液管道(B1)放入高盐溶液中,在system control窗口点击工具栏内的manual,选择pump→pump wash explorer,选中A1,B1管道为ON,execute。泵清洗将自动结束。(Fig 4) Fig 4、AKTA Explorer的泵清洗操作 2.4安装层析柱
蛋白质分离纯化的步骤
蛋白质分离纯化的一般程序可分为以下几个步骤: (一)材料的预处理及细胞破碎 分离提纯某一种蛋白质时,首先要把蛋白质从组织或细胞中释放出来并保持原来的天然状态,不丧失活性。所以要采用适当的方法将组织和细胞破碎。常用的破碎组织细胞的方法有: 1. 机械破碎法 这种方法是利用机械力的剪切作用,使细胞破碎。常用设备有,高速组织捣碎机、匀浆器、研钵等。 2. 渗透破碎法 这种方法是在低渗条件使细胞溶胀而破碎。 3. 反复冻融法 生物组织经冻结后,细胞内液结冰膨胀而使细胞胀破。这种方法简单方便,但要注意那些对温度变化敏感的蛋白质不宜采用此法。 4. 超声波法 使用超声波震荡器使细胞膜上所受张力不均而使细胞破碎。 5. 酶法 如用溶菌酶破坏微生物细胞等。 (二)蛋白质的抽提 通常选择适当的缓冲液溶剂把蛋白质提取出来。抽提所用缓冲液的pH、离子强度、组成成分等条件的选择应根据欲制备的蛋白质的性质而定。如膜蛋白的抽提,抽提缓冲液中一般要加入表面活性剂(十二烷基磺酸钠、tritonX-100 等),使膜结构破坏,利于蛋白质与膜分离。在抽提过程中,应注意温度,避免剧烈搅拌等,以防止蛋白质的变性。(三)蛋白质粗制品的获得选用适当的方法将所要的蛋白质与其它杂蛋白分离开来。比较方便的有效方法是根据蛋白质溶解度的差异进行的分离。常用的有下列几种方法: 1.等电点沉淀法不同蛋白质的等电点不同,可用等电点沉淀法使它们相互分离。 2.盐析法 不同蛋白质盐析所需要的盐饱和度不同,所以可通过调节盐浓度将目的蛋白沉淀析出。被盐析沉淀下来的蛋白质仍保持其天然性质,并能再度溶解而不变性。 3.有机溶剂沉淀法 中性有机溶剂如乙醇、丙酮,它们的介电常数比水低。能使大多数球状蛋白质在水溶液中的溶解度降低,进而从溶液中沉淀出来,因此可用来沉淀蛋白质。此外,有机溶剂会破坏蛋白质表面的水化层,促使蛋白质分子变得不稳定而析出。由于有机溶剂会使蛋白质变性,使用该法时,要注意在低温下操作,选择合适的有机溶剂浓度。 (四)样品的进一步分离纯化
洗瓶岗位标准操作规程
洗瓶岗位标准操作规程 目的: 建立粉针剂车间洗瓶岗位标准操作程序,使操作达到标准化、规范化,保证洗瓶质量。 2. 范围: 适用于粉针车间洗瓶岗位。 3.职责: 操作人员对本标准实施负责,车间主任、QA检查员负责监督检查。 4. 程序: 4.1. 操作前准备: 4.1.1. 更衣: 4.1.1.1. 理瓶操作人员按进出一般生产区更衣规程(SOP SC0009)进行更衣。 4.1.1.2. 洗瓶操作人员按进出十万级洁净区更衣规程(SOP SC0011)进行更衣。 4.1.2. 检查室内清场、清洁情况。 4.1.3. 将“清场合格证”附于批生产记录上。 4.1.4. 根据“生产指令”备抗生素瓶。 4.2. 操作过程: 4.2.1. 理瓶: 4.2.1.1. 根据“批生产指令”领取抗生素瓶。 去掉外包装箱,将中包装盒摆放在操作台上。 4.2.1.3. 打开中包装盒,剔出不良品,将瓶由上瓶窗口传入洗瓶间。 4.3. 洗瓶: 4.3.1. 检查纯化水、注射用水管路是否畅通,打开阀门并放水5分钟。 4.3.2. 洗瓶机:按超声波洗瓶机清洁规程(SOP SC6013)进行清洁。 4.3.3. 隧道烘箱:按隧道烘箱清洁规程(SOP SC0058)进行清洁。 4.3.4. 纯化水经0.22μm孔径筒式滤芯过滤,注射用水经0.22μm孔径筒式滤芯 过滤,压缩空气经0.01孔径滤芯过滤。 4.3. 5. 检查纯化水、注射用水澄明度,符合规定后,开机进行试车。 4.3.6. 在放水过程中检查注射用水澄明度,如不符合规定立即停车,将水排放一段 时间后,再次检查仍不合格,则需更换滤芯,直至检查合格。
蛋白纯化的一般原则及方法选择
随着分子生物学的发展,越来越多的科研人员熟练掌握了分子生物学的各种试验技术,并研制成套试剂盒,使基因克隆表达变得越来越容易lIl。但分子生物学的上游工作往往并非是最终目的,分子克隆与表达的关键是要拿到纯的表达产物,以研究其生物学作用,或者大量生产出可用于疾病治疗的生物制品。相对与上游工作来说,分子克隆的下游工作显得更难,蛋白纯化工作非常复杂,除了要保证纯度外,蛋白产品还必须保持其生物学活性。纯化工艺必须能够每次都能产生相同数量和质量的蛋白,重复性良好。这就要求应用适应性非常强的方法而不是用能得到纯蛋白的最好方法去纯化蛋白。在实验室条件下的好方法却可能在大规模生产应用中失败,因为后者要求规模化,且在每日的应用中要有很好的重复性。本文综述了蛋白质纯化的基本原则和各种蛋白纯化技术的原理、优点及局限性,以期对蛋白纯化的方法选择及整体方案的制定提供一定的指导。 1 蛋白纯化的一般原则 蛋白纯化要利用不同蛋白间内在的相似性与差异,利用各种蛋白间的相似性来除去非蛋白物质的污染,而利用各蛋白质的差异将目的蛋白从其他蛋白中纯化出来。每种蛋白间的大小、形状、电荷、疏水性、溶解度和生物学活性都会有差异,利用这些差异可将蛋白从混合物如大肠杆菌裂解物中提取出来得到重组蛋白。蛋白的纯化大致分为粗分离阶段和精细纯化阶段二个阶段。粗分离阶段主要将目的蛋白和其他细胞成分如DNA、RNA等分开,由于此时样本体积大、成分杂,要求所用的树脂高容量、高流速,颗粒大、粒径分布宽.并可 以迅速将蛋白与污染物分开,防止目的蛋白被降解。精细纯化阶段则需要更高的分辨率,此阶段是要把目的蛋白与那些大小及理化性质接近的蛋白区分开来,要用更小的树脂颗粒以提高分辨常用的离子交换柱和疏水柱,应用时要综合考虑树脂的选择性和柱效两个因素。选择性指树脂与目的蛋白结合的特异性,柱效则是指蛋白的各成分逐个从树脂上集中洗脱的能力,洗脱峰越窄,柱效越好。仅有好的选择性,洗脱峰太宽,蛋白照样不能有效分离。 2.各种蛋白纯化方法及优缺点 2.1蛋白沉淀蛋白能溶于水是因为其表面有亲水性氨基酸。在蛋白质的等电点处若溶液的离子强度特别高或特别低,蛋白则倾向于从溶液中析出。硫酸铵是沉淀蛋白质最常用的盐,因为它在冷的缓冲液中溶解性好,冷的缓冲液有利于保护蛋白的活性。硫酸铵分馏常用做纯化的第一步,它可以初步粗提蛋白质,去除非蛋白成分。蛋白质在硫酸铵沉淀中较稳定,可以短期在这种状态下保存中间产物,当前蛋白质纯化多采用这种办法进行粗分离翻。在规模化生产上硫酸铵沉淀方法仍存在一些问题,硫酸铵对不锈钢器具的腐蚀性很强。其他的盐如硫酸钠不存在这种问题,但其纯化效果不如硫酸铵。除了盐析外蛋白还可以用多聚物如PEG 和防冻剂沉淀出来,PEG是一种惰性物质,同硫酸铵一样对蛋白有稳定效果, 在缓慢搅拌下逐渐提高冷的蛋白溶液中的PEG浓度,蛋白沉淀可通过离心或过滤获得,蛋白可在这种状态下长期保存而不损坏。蛋白沉淀对蛋白纯化来说并不是多么好的方法,因为它只能达到几倍的纯化效果,而我们在达到目的前需要上千倍的纯化。其好处是可以把蛋白从混杂有蛋白酶和其他有害杂质的培养基及细胞裂解物中解脱出来。
蛋白纯化的基本思路
蛋白质的提取和纯化-- 选择分离材料及预处理蛋白质的提取和纯化-- 选择分离材料及预处理 以蛋白质和结构与功能为基础,从分子水平上认识生命现象,已经成为现代生物学发展的主要方向,研究蛋白质,首先要得到高度纯化并具有生物活性的目的物质。 蛋白质的制备工作涉及物理、化学和生物等各方面知识,但基本原理不外乎两方面。一是得用混合物中几个组分分配率的差别,把它们分配到可用机械方法分离的两个或几个物相中,如盐析,有机溶剂提取,层析和结晶等;二是将混合物置于单一物相中,通过物理力场的作用使各组分分配于来同区域而达到分离目的,如电泳,超速离心,超滤等。在所有这些方法的应用中必须注意保存生物大分子的完整性,防止酸、硷、高温,剧烈机械作用而导致所提物质生物活性的丧失。 蛋白质的制备一般分为以下四个阶段:选择材料和预处理,细胞的破碎及细胞器的分离,提取和纯化,浓细、干燥和保存。 微生物、植物和动物都可做为制备蛋白质的原材料,所选用的材料主要依据实验目的来确定。对于微生物,应注意它的生长期,在微生物的对数生长期,酶和核酸的含量较高,可以获得高产量,以微生物为材料时有两种情况:( 1 )得用微生物菌体分泌到培养基中的代谢产物和胞外酶等;(2)利用菌体 含有的生化物质,如蛋白质、核酸和胞内酶等。植物材料必须经过去壳,脱脂并注意植物品种和生长发育状况不同,其中所含生物大分子的量变化很大,另外与季节性关系密切。对动物组织,必须选择有效成份含量丰富的脏器组织为原材料,先进行绞碎、脱脂等处理。另外,对预处理好的材料,若不立即进行实验,应冷冻保存,对于易分解的生物大分子应选用新鲜材料制备。 细胞的破碎 1、高速组织捣碎:将材料配成稀糊状液,放置于筒内约1/3 体积,盖紧筒盖,将调速器先拨至最慢处, 开动开关后,逐步加速至所需速度。此法适用于动物内脏组织、植物肉质种子等。 2、玻璃匀浆器匀浆:先将剪碎的组织置于管中,再套入研杆来回研磨,上下移动,即可将细胞研碎,此法细胞破碎程度比高速组织捣碎机为高,适用于量少和动物脏器组织。 3、超声波处理法:用一定功率的超声波处理细胞悬液,使细胞急剧震荡破裂,此法多适用于微生物材料, 用大肠杆菌制备各种酶,常选用50-100 毫克菌体/毫升浓度,在1KG 至10KG 频率下处理10-15 分钟,此法的缺点是在处理过程会产生大量的热,应采取相应降温措施。对超声波敏感和核酸应慎用。 4、反复冻融法:将细胞在-20 度以下冰冻,室温融解,反复几次,由于细胞内冰粒形成和剩余细胞液的盐浓度增高引起溶胀,使细胞结构破碎。 5、化学处理法:有些动物细胞,例如肿瘤细胞可采用十二烷基磺酸钠(SDS)、去氧胆酸钠等细胞膜破 坏,细菌细胞壁较厚,可采用溶菌酶处理效果更好。 无论用哪一种方法破碎组织细胞,都会使细胞内蛋白质或核酸水解酶释放到溶液中,使大分子生物 降解,导致天然物质量的减少,加入二异丙基氟磷酸(DFP)可以抑制或减慢自溶作用;加入碘乙酸可以
称量岗位标准操作规程
XXXXXXX有限公司岗位清洁标准操作规程 1目的:建立称量岗位清洁标准操作规程 2范围:适用称量岗位清洁标准操作。 3 责任者:由岗位操作人员、车间管理人员负责、质量监督员监督。 4 依据:《药品生产质量管理规范》(2010年修订)。 5 内容: 5.1 清洁条件 5.1.1收集的残留物料进行收集处理,操作现场的所有物料转出操作间。 5.2 清洁工具 5.2.1洁净抹布、塑料水桶、笤帚(不脱落)、簸箕、塑料刷、拖布(不脱落)。 5.3 清洁剂和消毒剂 5.3.1饮用水、纯化水、84消毒液、0.2%新洁尔灭、75%乙醇溶液。 5.4 清洁频次、要求 5.4.1 同品种、批次连续生产时可进行局部清洁。 5.4.2 更换品种、批次时必须进行彻底清洁。 5.4.3 设备按各设备清洁标准操作规程清洁结束后必须表面光亮、无异物。5.4.4 操作间清洁后必须干净、无污迹。 5.4.5 对设备接触物料的部位用75﹪酒精擦拭消毒。 6 清洁程序和方法 6.1 将设备、地面撒落的物料、无效的标识卡进行清理,置废弃物容器内。 6.2 清洁顶棚、墙面及门。
称量岗位清洁标准操作规程共2 页第2 页 6.2.1 用洁净抹布分别擦拭顶棚、灯罩、墙面、门至无污迹、无水痕。 6.3 清洁操作台、记录桌 6.3.1 用洁净抹布擦拭至无污迹、无水痕; 6.3.2 用洁净抹布将文件夹表面进行擦拭,保护岗位文件不受损坏。 6.4 清洁地面、圆弧角 6.4.1 清扫地面杂物收集置废弃物桶。 6.4.2 用洁净拖布,挤压去水,擦拭地面至无污迹; 6.4.3 用消毒液浸泡拖布并挤压去水后将地面擦拭、消毒。 7 清场确认 7.1 清场结束后先自查,自查合格经QA确认后发放清场合格证并挂“已清洁”标 识。 7.2 填写清场记录,清场合格证正本附本批生产记录中,副本留在现场。 8 消毒液的配制 8.1 配制方法依据《清洁剂与消毒剂的配制标准操作规程》进行配制。 8.2 消毒液隔月交替使用。 9 清洁有效期:本次清洁结束至下次生产前的时间间隔为24小时,超过有效期则进行重新清场并消毒。 10 工器具清洁 10.1 所有工器具置容器洗涤间清洁,清洁完毕待确认后挂“已清洁”状态标识。 11 洁具的清洁和存放:依照《清洁用具的使用、清洁、存放标准操作规程》进 行
GE NOVAGEN 镍柱纯化系统流程
蛋白纯化系统操作流程 一、蛋白的诱导:蛋白原核表达 1、取菌种接种于含Amp LB固体培养基中(分区划线),37℃培养过夜; 2、挑取单克隆接种于5ml含Amp LB液体培养基中,37℃振摇过夜; 3、从过夜培养物中取2ml接种于100ml Amp LB液体培养基中,振摇2h(留样1ml); 4、加入一定终浓度IPTG,37℃诱导表达4h(留样1ml),离心,弃上清收集细菌。 存入4℃。 二、蛋白表达状态分析(可溶性or包涵体表达) 取少量(1ml)诱导菌体沉淀,加入不含变性剂(如盐酸胍,尿素等)PBS(150μl),超声裂解。分离上清和沉淀,分别SDS-PAGE电泳。 三、蛋白的纯化 纯化前准备 1.推荐在中性至弱碱性条件下(pH 7-8)结合重组蛋白。磷酸盐buffer是常用的缓冲液, Tric-Cl在一般情况下可用,但要注意它会降低结合强度。 2.避免在buffer中包含EDTA或柠檬酸盐等螯合剂 3.若重组蛋白以包涵体形式表达,在所有的buffer中添加6 M 盐酸胍或8 M 尿素 注: 1.包含尿素的样品可直接进行SDS-PAGE分析,若样品中包含盐酸胍,在SDS-PAGE前则 需先用含有尿素的buffer进行透析 2.除利用咪唑洗脱蛋白,其它方法,如低pH 值法等可被应用,详见说明书 Bingding buffer 中咪唑的浓度 在洗涤时所用的Bingding buffer 中咪唑浓度越大,重组蛋白纯度越高。但过高的咪唑浓度将导致蛋白的洗脱。合适的咪唑浓度需要优化。 Buffer 的准备
所用的水及化学物质须是高纯度的。Buffer 在使用前需经0.45 μm滤膜过滤 所用高纯度的咪唑需在280nm 处无吸光度或吸光度极低 推荐buffer Bingding buffer:20 mM 磷酸盐 0.5 M NaCl 20- 40 mM 咪唑pH 7.4 (咪唑浓度是蛋白依赖的,可变!)Elution buffer :20 mM 磷酸盐 0.5 M NaCl 500 mM 咪唑pH 7.4 (咪唑浓度是蛋白依赖的,可变!) 样品准备 样品需被充分溶解。过柱前经0.45 μm滤膜过滤。样品以pH 7.4 binding buffer 溶解。勿用强酸强碱调节pH 值,否则将可能导致沉淀。 重力纯化法Ni-NTA Column 准备 1. 温和地颠倒瓶中的Ni-NTA Agarose 数次。 2. 吸取2ml的树脂加入15ml离心管中,使树脂在重力(5–10 minutes)或低速离心(5 minute at 500 × g),轻柔的吸出上清。 3. 加入5ml的无菌蒸馏水,温和的颠倒柱子3min,离心5 minute at 500 × g,轻柔的吸出上清。 4. 用bingding buffer 重复第3步。 5. 在Ni 柱中加入等体积的bingding buffer,制成50%的slurry 样品与Ni 柱结合 1.每1ml 50%的slurry中加入4ml 的样品。1ml 50%的slurry 可结合20mg His-蛋白 2.将混合物室温,低速振荡孵育1h Buffer 洗涤及洗脱 1.离心5 minute at 500 × g,轻柔的吸出上清。上清保存放在4℃for SDS-PAGE
制粒岗位标准操作规程
XXXXXXXX有限公司岗位清洁标准操作规程 1 目的:建立制粒岗位清洁标准操作规程。 2 范围:适用于制粒岗位清洁标准操作。 3 责任者:由岗位操作人员、车间管理人员负责、质量监督员监督。 4 依据:《药品生产质量管理规范》(2010年修订)。 5 内容: 5.1 清洁条件 5.1.1收集的残留物料进行收集处理,操作现场的所有物料转出操作间。 5.2 清洁工具 5.2.1洁净抹布、塑料水桶、笤帚(不脱落)、簸箕、塑料刷、拖布(不脱落)。 5.3 清洁剂和消毒剂 5.3.1饮用水、纯化水、84消毒液、0.2%新洁尔灭、75%乙醇。 5.4 清洁频次、要求 5.4.1 同品种、批次连续生产时可进行局部清洁。 5.4.2 更换品种、批次时必须进行彻底清洁。 5.4.3 设备按各设备清洁标准操作规程清洁结束后必须表面光亮、无异物。5.4.4 操作间清洁后必须干净、无污迹。 5.4.5 对设备接触物料部位用75﹪乙醇擦拭消毒。 6 清洁程序和方法 6.1 将设备、地面撒落的物料、无效的标识卡进行清理,置废弃物容器内。6.2 清洁顶棚、墙面及门。
制粒岗位清洁标准操作规程共 2 页第 2 页 6.2.1 用洁净抹布分别擦拭顶棚、灯罩、墙面、门至无污迹。 6.2.2 用消毒液将顶棚、墙面及门全面擦拭消毒。 6.3 摇摆式制粒机的清洁依据《摇摆式制粒机清洁标准操作规程》进行清洁。 槽型混合机的清洁依据《槽型混合机清洁标准操作规程》进行清洁。 6.4 清洁操作台、记录桌 6.4.1 用洁净抹布擦拭至无污迹、无水痕; 6.4.2 用洁净抹布将文件夹表面进行擦拭,保护岗位文件不受损坏。 6.5 清洁地面、圆弧角 6.5.1 清扫地面杂物收集置废弃物桶。 6.5.2 用洁净拖布,挤压去水,擦拭地面至无污迹; 6.5.3 用消毒液浸泡拖布并挤压去水后将地面擦拭、消毒。 7 清场确认 7.1 清场结束后先自查,自查合格经QA确认后发放清场合格证并挂“已 清洁”标识。 7.2 填写清场记录,清场合格证正本附本批生产记录中,副本留在现场。 8 消毒液的配制 8.1 配制方法依据《清洁剂与消毒剂的配制标准操作规程》进行配制。 8.2 消毒液隔月交替使用。 9 清洁有效期:本次清洁结束至下次生产前的时间间隔为24小时,超过有效期则进行重新清场并消毒。 10 工器具清洁 10.1 所有工器具置容器洗涤间清洁,清洁完毕待确认后挂“已清洁”状态标识。 11 洁具清洁和存放:依照《清洁用具的使用、清洁、存放标准操作规程》进行。
Protocol蛋白质纯化步骤
Protocol 蛋白质纯化方法(镍柱) 柱前操作 1.IPTG诱导后,收菌,8000rpm/min(r/m)离心10min; 2.用Binding Buffer(BB)溶解(每100ml原菌液加BB 20ml),超声裂解30min(工作:5s,停止:5s),1500r/m离心10min,去除杂质; 3.取上清,12000r/m离心20min, 得包涵体; 4.用含2M尿素的BB洗包涵体,12000r/m离心20min,(上清做电泳);??? 5.用含6M尿素的BB溶解包涵体,12000r/m离心20min,(上清做电泳); 6.对照电泳结果,将上清或包涵体溶解液上柱; 平衡柱子(柱体积:V) 7. 3V(3倍柱体积)ddH2O(洗乙醇); 8. 5V Charge Buffer(CB); ??? 9. 3V BB; 柱层析 10.上样; 11. 10V Washing Buffer(WB); 12. 6V Elute Buffer(EB); 13.分管收集,每管1~2ml. 各种缓冲液配方 1. 8×BB: 4M NaCl, 160mM Tris-HCl, 40mM imidazole(咪唑),pH=7.9 1000ml NaCl: 58.44×4=233.76g Tris-HCl: 121.14×160×10-3=19.3824g Imidazole: 68.08×40×10-3=2.7232g 2. 8×CB: 400mM NiSO4 1000ml NiSO4: 262.8×400×10-3=105.12g 3. 8×WB: 4M NaCl, 160mM Tris-HCl, 480mM imidazole, pH=7.9 1000ml NaCl: 233.76g, Tris-HCl:19.3824g, Imidazole: 32.6784g 4. 4×EB: 2M NaCl, 80mM Tris-HCl, 4M imidazole, pH=7.9 1000ml NaCl: 118.688g, Tris-HCl:9.6912g, Imidazole: 272.32g 5. 6M 尿素 1000ml 尿素:60.06×6=360.36g
蛋白纯化系统Biologic-LP使用说明
蛋白纯化系统Biologic-LP使用说明 Biologic-LP是蛋白质层析纯化系统, 其原理是利用不同蛋白分子所具有的特性(如等电点、分子量及亲水或疏水性)与层析柱中的介质产生的吸附作用后,再用相应的洗脱液来对吸附在层析柱上的蛋白进行洗脱。根据目标蛋白及不同层析柱介质的特性,设计相应的洗脱程序可以使目标蛋白与其他杂蛋白先后从层析柱上洗脱下来。通过观察紫外光的吸收峰,可分别收集不同时段洗脱下来的蛋白液。蛋白混合物通过这样的程序可被分离至单个蛋白。通常分布在混合物中的目标蛋白需要通过组合而不是单一的层析路线来进行分离操作。常规的分离路线如通过疏水层析—离子交换—疏水层析的技术路线来有效分离目标蛋白。 本层析系统使用主要分为三个部分。首先在使用前确认分离的技术路线和使用的层析柱。其次根据层析柱使用的要求配制相关试剂和确定层析过程的参数。最后通过层析操作分离纯化目标蛋白,并清洗层析柱和管道以确保仪器能长期有效使用。 一设计蛋白的纯化路线及选择不同的层析柱及层析方法根据目标蛋白的特性及来源,设计纯化的路线并确定每一步操作所需要的层析柱及层析方法。根据不同层析方法的要求,准备蛋白样品及洗脱液及洗脱方式(如线形洗脱或梯度洗脱)。而后确认层析操作中的主要参数。
二层析系统的操作 以下是对所有层析操作中共同的步骤进行的描述。特别注意的是不同的分离方式如离子交换和疏水层析它们的原理和参数设置完全不同。这里仅就相同的操作进行描述,具体的参数设置见使用说明书并咨询负责本仪器的老师,切不可擅自操作,以免破坏仪器。 1、确定目标蛋白层析柱的选择,不同的分离方式选择不同的层析柱。 2、样品制备。根据层析柱介质对蛋白样品的要求,制备样品和洗脱 液。所有用于层析的溶液及样品均要通过0.45μm膜过滤,以免堵塞层析柱。 3、打开层析仪电源,按照显示屏的提示,分别设置好A液、B液、 流速、时间等相关参数,并将接样管插入接样仪。 4、打开电脑及Biologic-LP Data View软件,观察层析过程是否正常 或是否需要调整,做好接样前的准备。 三、层析系统的维护 操作结束后,按仪器使用说明,清洗层析柱及管道,将层析柱保存好,备用。特别注意不同的层析柱要求的清洗方式不同,对管道的清洗也不同,层析柱的保存方式也不同。清洗和保存时一定要按照使用说明书的要求进行操作,不能出现错误以免对层析系统造成破坏。
生产用培养基制备岗位标准操作规程
黑龙江正康生物技术股份有限公司GMP管理文件 一、目的:为保证培养基能够满足生产要求,特定培养基制备岗位操作规程。 二、适用范围:适用于培养基制备岗位的操作。 三、责任者:分发部门负责人,质量监督员,工艺员,操作人员。 四、正文: 1 操作前准备: 1.1 接收指令,根据指令情况填写生产状态标识牌,标明当日生产产品的品名、规格、批号、批量、岗位名称、生产日期。 1.2 检查上批次清场情况,有上一批次生产“清场合格证”,并确认无上次生产遗留物。 1.3 检查生产现场卫生状况,有厂房“已清洁”状态标识,并在效期内。 1.4 检查设备、设施及状态情况,有设备“已清洁”状态标识,并在效期内。 1.5 检查容器具卫生状况,有容器具“已清洁”状态标识,并在效期内。 1.6 检查生产用衡器是否处于水平状态,是否归零,是否有检定合格证,并在有效期内。 1.7 检查确认批生产记录及相应的配套记录已准备齐全。 2 生产操作: 2.1 领料: 2.1.1 根据生产指令开出限额领料单,领取所需用的原辅材料,并有质量管理部的检验合格报告
单。检查报告单上的物料编码、品名、数量、规格及外观质量,合格后才能使用;如不符合要求应拒绝收料; 2.1.2 按照《物料进入生产区标准操作规程》清除原辅料外包装物表面的异物灰尘,并清洁消毒,再复核物料编码、品名,规格、检查外观、数量,避免差错,放入物料暂存间中待用。 2.2 称量: 2.2.1 称量前应核对原辅料物料编码、品名、批号、生产厂家、规格等,应与检验报告单相符。 2.2.2 原辅料的使用量应根据原料的实际含量、含水量等因素进行换算,按检验用培养基制配法或各产品的工艺规程所规定的量进行100%的投料。 2.2.3 称量时所用的取料铲必须专用,不能混用,数量应有复核人,操作人和复核人均应在称量原始记录上签名。 2.2.4 液体药品临用前单独称量,称量完毕要立即清理称量台和台秤。 2.2.5 剩余的原辅料应密封贮存,并在容器外标明物料编码、品名、批号、日期、剩余量及使用人。 2.3 配制: 2.3.1 注意事项: 2.3.1.1 根据规程,核对所用原辅料的物料编码、品名、批号、生产厂、规格等; 2.3.1.2 每一个配制器皿必须标明所配培养基的品名、规格、批号和配制量; 2.3.1.3 配制时,每一种原辅料的加入和调制必须由核对人确认并作好记录; 2.3.1.4 配制过程中的温度调节和配制的最后定量均要有复核人确认,并有操作人和复核人签字。 2.3.2 配制过程 2.3.2.1 使用市售的成品培养基的配制过程按使用说明书配制即可。 2.3.2.2 使用各种原材料配制的配制过程见附注。
蛋白质纯化的方法选择
蛋白质纯化的方法选择 随着分子生物学的发展,越来越多的科研人员熟练掌握了分子生物学的各种试验技术,并研制成套试剂盒,使基因克隆表达变得越来越容易。但分子生物学的上游工作往往并非是最终目的,分子克隆与表达的关键是要拿到纯的表达产物,以研究其生物学作用,或者大量生产出可用于疾病治疗的生物制品。相对与上游工作来说,分子克隆的下游工作显得更难,蛋白纯化工作非常复杂,除了要保证纯度外,蛋白产品还必须保持其生物学活性。纯化工艺必须能够每次都能产生相同数量和质量的蛋白,重复性良好。这就要求应用适应性非常强的方法而不是用能得到纯蛋白的最好方法去纯化蛋白。在实验室条件下的好方法却可能在大规模生产应用中失败,因为后者要求规模化,且在每日的应用中要有很好的重复性。本文综述了蛋白质纯化的基本原则和各种蛋白纯化技术的原理、优点及局限性,以期对蛋白纯化的方法选择及整体方案的制定提供一定的指导。 1、蛋白纯化的一般原则 蛋白纯化要利用不同蛋白间内在的相似性与差异,利用各种蛋白间的相似性来除去非蛋白物质的污染,而利用各蛋白质的差异将目的蛋白从其他蛋白中纯化出来。每种蛋白间的大小、形状、电荷、疏水性、溶解度和生物学活性都会有差异,利用这些差异可将蛋白从混合物如大肠杆菌裂解物中提取出来得到重组蛋白。蛋白的纯化大致分为粗分离阶段和精细纯化阶段二个阶段。粗分离阶段主要将目的蛋白和其他细胞成分如DNA、RNA等分开,由于此时样本体积大、成分杂,要求所用的树脂高容量、高流速,颗粒大、粒径分布宽.并可以迅速将蛋白与污染物分开,防止目的蛋白被降解。精细纯化阶段则需要更高的分辨率,此阶段是要把目的蛋白与那些大小及理化性质接近的蛋白区分开来,要用更小的树脂颗粒以提高分辨率,常用离子交换柱和疏水柱,应用时要综合考虑树脂的选择性和柱效两个因素。选择性树脂与目的蛋白结合的特异性,柱效则是指各蛋白成分逐个从树脂上集中洗脱的能力,洗脱峰越窄,柱效越好。仅有好的选择性,洗脱峰太宽,蛋白照样不能有效分离。 2、各种蛋白纯化方法及其优、缺点 2.1 蛋白沉淀蛋白能溶于水是因为其表面有亲水性氨基酸,在蛋白质的等电点处若溶液的离子强度特别高或者特别低,蛋白则倾向于从溶液中析出。硫酸铵是沉淀蛋白最常用的盐,因为它在冷的缓冲液中溶解性好,冷的缓冲液有利于保持目的蛋白的活性。硫酸铵分馏常用作试验室蛋白纯化的第一步,它可以初步粗提蛋白质,去除非蛋白成分。蛋白质在硫酸铵沉淀中较稳定,可以短期在这种状态下保存中间产物,当前蛋白质纯化多采用这种办法进行粗分离翻。在规模化生产上硫酸铵沉淀方法仍存在一些问题,硫酸铵对不锈钢器具的腐蚀性很强。其他的盐如硫酸钠不存在这种问题,但其纯化效果不如硫酸铵。除了盐析外蛋白还可以用多聚物如PEG和防冻剂沉淀出来,PEG是一种惰性物质,同硫酸铵一样对蛋白有稳定效果,在缓慢搅拌下逐渐提高冷的蛋白溶液中的PEG浓度,蛋白沉淀可通过离心或过滤获得,蛋白可在这种状态下长期保存而不损坏。蛋白沉淀对蛋白纯化来说并不是多么好的方法,因为它只能达到几倍的纯化效果,而我们在达到目的前需要上千倍的纯化。其好处是可以把蛋白从混杂有蛋白酶和其他有害杂质的培养基及细胞裂解物中解脱出来。 2.2 缓冲液的更换虽然更换缓冲液不能提高蛋白纯度,但它却在蛋白纯化方案中起着极其重要的作用。不同的蛋白纯化方法需要不同pH及不同离子强度的缓冲液。假如你用硫酸铵将蛋白沉淀出来,毫无疑问蛋白是处在高盐环境中,需要想办法脱盐,可用的方法有利用半透膜透析,通过勤换透析液体去除盐分,此法尚可,但需几个小时,通常要过夜,也难以用于大规模纯化中。新型的设备将透析膜夹在两个板中间,板的一侧加缓冲液,另一侧加需脱盐的蛋白溶液,并在蛋白溶液一侧通过泵加压,可以使两侧溶液在数小时内达到平衡,若增加对蛋白溶液的压力,还可迫使水分和盐更多通过透析膜进入透析液达到对蛋白浓缩的目的。也有出售的脱盐柱,柱内的填料是小孔径的颗粒,蛋白分子不能进入孔内,先于高浓度盐离子从柱中流出,从而使二者分离。蛋白纯化的每一步都会造成目的蛋白的丢失,缓冲液平衡的步骤尤甚。蛋白会结合在任何它能接触的表面上,剪切力、起泡沫和离子强度的快速变化很容易让蛋白失活。 2.3 离子交换色谱这是在所有的蛋白纯化与浓缩方法中最有效方法。基于蛋白与离子交换树脂间的相互电荷作用,通过选择不同的缓冲液,同一种蛋白既可以和阴离子交换树脂(能结合带负电荷的分子)结合,也可以和阳离子交换树脂结合。树脂所用的带电基团有四种:二乙基氨基乙基用于弱的阴离子交换树脂;羧甲基用于弱的阳离子交换树脂;季铵用于强阴离子交换树脂;甲基磺酸酯用于强阳离子交换树脂。蛋白质由氨基酸组成,氨基酸在不同的pH环境中所带总电荷不同。大多数蛋白在生理pH(pH6~8)下带负电荷,需用阴离子交换柱纯化,极端的pH下蛋白会变性失活.应尽量避免。由于在某个特定的pH下不同的蛋白所带电荷数不同,与树脂的结合力也不同,随着缓冲液中盐浓度的增加或pH的变化,蛋白按结合力的强弱被依次洗脱。在工业化生产中更多地是改变盐浓度而不是去改变pH值,因为前者更容易控制。在实验室中几乎总是用盐浓度梯度去洗脱离子交换柱,利用泵的辅助可以使流入柱的缓冲液中盐浓度平稳地上升,当离子强度能够中和蛋白的电荷时,蛋白就被从柱上洗脱下来。但在工业生产中盐浓度很难精确控制,所以常用分步洗脱而不足连续升高的盐梯度。与排阻层析相比,离子交换特异性更好,有更多的参数可以调整以获得最优的纯化效果,树脂也比较便宜。值得一提的是,即便是用最精确控制的条件,仅用离子交换单一的方法也得不到纯的蛋白,还需要其他的纯化步骤。
02(改动)纯化水岗位标准操作规程
纯化水岗位标准操作规程 1. 目的: 建立纯化水岗位标准操作规程,使其操作标准化、规范化。 2. 范围:适用于纯化水岗位的操作。 3. 责任:车间主任、质监员、纯化水岗位操作工对本标准的实施负责。 4. 内容: 包括纯化水的生产操作方法和要点,工艺用水的质量标准及控制,安全和劳动保护,异常情况处理和报告等。 4.1纯化水的生产操作方法和要点: 4.1.1纯化水制备工艺流程如下: 饮用水石英砂过滤器活性炭过滤器精密过滤器 二级高压泵pH调节装置中间贮罐一级反渗透阻垢剂投加装置一级高压泵 二级反渗透微滤各使用点 4.1.2 纯化水制备按照纯化水制备设备操作规程进行开机操作。 4.1.3控制反渗透膜的进水流量和压力,一级反渗透产水量达到24T/h,浓水排放量为8T/h左右,一级产水电导率在20μs/cm以下方可进入中间水箱;二级反渗透产水量达到20T/h,浓水排放量为4T/h左右,二级产水电导率在2μs/cm以下,方可进入纯化水罐。 ----------------------------精品word文档值得下载值得拥有 ----------------------------------------------
4.1.4 纯化水的输送:打开纯化水泵进水阀、出口阀,开启纯化水泵和紫外灯杀菌器,然后缓慢打开精密过滤器进水阀及排气阀,精密过滤器内空气排完后关闭排气阀,再打开纯化水循环系统各阀门,进行循环保压0.3MPa以上,供各使用点使用。 4.1.5阻垢剂的配制方法:用2份阻垢剂原液、8份反渗透产水,配成20%的阻垢剂溶液。每次所配溶液应在一周内用完,夏季温度较高,每次更换药液时应彻底清洗或消毒药箱。计量泵的启停,应与反渗透机组同步。 4.1.6 PH调节装置:调节碱液进入速度有效除去一级产水中的二氧化碳,使二级产水电导率在2μs/cm以下。 碱液配制:氢氧化钠液浓度为2.5%。取1kg氢氧化钠(化学纯、500g/瓶)加入40L碱箱中,加纯化水至40L,搅拌至全部溶解备用。 4.2 纯化水的质量标准及控制 4.2.1纯化水质量标准 纯化水为蒸馏法、离子交换法、反渗透法或其他适宜的方法制得的供药用的水,不含任何附加剂。 标准依据:《中国药典》2005年版二部第303页,见下表: ----------------------------精品word文档值得下载值得拥有 ----------------------------------------------
蛋白质的分离纯化方法(参考资料)
蛋白质的分离纯化方法 2.1根据分子大小不同进行分离纯化 蛋白质是一种大分子物质,并且不同蛋白质的分子大小不同,因此可以利用一些较简单的方法使蛋白 质和小分子物质分开,并使蛋白质混合物也得到分离。根据蛋白质分子大小不同进行分离的方法主要有透析、超滤、离心和凝胶过滤等。透析和超滤是分离蛋白质时常用的方法。透析是将待分离的混合物放入半透膜制成的透析袋中,再浸入透析液进行分离。超滤是利用离心力或压力强行使水和其它小分子通过半透膜,而蛋白质被截留在半透膜上的过程。这两种方法都可以将蛋白质大分子与以无机盐为主的小分子分开。它们经常和盐析、盐溶方法联合使用,在进行盐析或盐溶后可以利用这两种方法除去引入的无机盐。由于超滤过程中,滤膜表面容易被吸附的蛋白质堵塞,以致超滤速度减慢,截流物质的分子量也越来越小。所以在使用超滤方法时要选择合适的滤膜,也可以选择切向流过滤得到更理想的效果离心也是经常和其它方法联合使用的一种分离蛋白质的方法。当蛋白质和杂质的溶解度不同时可以利用离心的方法将它们分开。例如,在从大米渣中提取蛋白质的实验中,加入纤维素酶和α-淀粉酶进行预处理后,再用离心的方法将有用物质与分解掉的杂质进行初步分离[3]。使蛋白质在具有密度梯度的介质中离心的方法称为密度梯度(区带)离心。常用的密度梯度有蔗糖梯度、聚蔗糖梯度和其它合成材料的密度梯度。可以根据所需密度和渗透压的范围选择合适的密度梯度。密度梯度离心曾用于纯化苏云金芽孢杆菌伴孢晶体蛋白,得到的产品纯度高但产量偏低。蒋辰等[6]通过比较不同密度梯度介质的分离效果,利用溴化钠密度梯度得到了高纯度的苏云金芽孢杆菌伴孢晶体蛋白。凝胶过滤也称凝胶渗透层析,是根据蛋白质分子大小不同分离蛋白质最有效的方法之一。凝胶过滤的原理是当不同蛋白质流经凝胶层析柱时,比凝胶珠孔径大的分子不能进入珠内网状结构,而被排阻在凝胶珠之外,随着溶剂在凝胶珠之间的空隙向下运动并最先流出柱外;反之,比凝胶珠孔径小的分子后流出柱外。目前常用的凝胶有交联葡聚糖凝胶、聚丙烯酰胺凝胶和琼脂糖凝胶等。在甘露糖蛋白提纯的过程中使用凝胶过滤方法可以得到很好的效果,纯度鉴定证明产品为分子量约为32 kDa、成分是多糖∶蛋白质(88∶12)、多糖为甘露糖的单一均匀糖蛋白[1]。凝胶过滤在抗凝血蛋白的提取过程中也被用来除去大多数杂蛋白及小分子的杂质[7]。 2.2 根据溶解度不同进行分离纯化 影响蛋白质溶解度的外部条件有很多,比如溶液的pH值、离子强度、介电常数和温度等。但在同一条件下,不同的蛋白质因其分子结构的不同而有不同的溶解度,根据蛋白质分子结构的特点,适当地改变外部条件,就可以选择性地控制蛋白质混合物中某一成分的溶解度,达到分离纯化蛋白质的目的。常用的方法有等电点沉淀和pH值调节、蛋白质的盐溶和盐析、有机溶剂法、双水相萃取法、反胶团萃取法等。 等电点沉淀和pH值调节是最常用的方法。每种蛋白质都有自己的等电点,而且在等电点时溶解度最
AKTA层析仪使用操作规程(AKTA蛋白纯化系统经典培训资料)
?KTApurifier层析仪使用操作规程 1、开机 打开仪器的主电源,打开电脑电源。待仪器自检完毕(CU950上面的3个指示灯完全点亮并不闪烁)。双击桌面上UNICORN图标,进入操作界面。 2、准备工作溶液和样品 所有的工作溶液和样品必须经过0.45μm的滤膜过滤,样品也可高速离心后取上清备用。当缓冲液中含有有机溶剂(如乙腈、甲醇),需在使用前用低频超声脱气10min。 3、清洗及管道准备 首先将A1管道放入缓冲液或平衡液,binding buffer中,将B1管道放入高盐溶液中或elution buffer,在system control窗口点击工具栏内的manual,选择 pump→pump wash basic,选中A1,B1管道为ON, execute。泵清洗将自动结束。 4、安装层析柱 在manual里选择pump→flow rate,输入流速1ml/ml,insert;选择Alarm&mon→alarm pressure,设置high alarm(输入填料的耐受压力,可在填料说明书中查到),insert,execute。待InjectionValve的1号位管道流出水后接入柱子的柱头,稍微拧紧后将柱下端的堵头卸掉接入管道连上紫外流动池。 5、开始纯化: 1)等待柱子平衡好了(观察电导COND,pH的数值和变化趋势)就准备上样了。此时将紫外调零,选择Alarm&mon→autozero,exectue。 2)用样品环上:将样品吸进注射器,推掉气泡,从injectionValve的3号位推入(进样量不得低于样品环的体积的2倍)推好后不要取下注射器。在manual里选择flowpath→injectionValve→inject,execute。 3)用泵上样:点击pause,将A1放入样品中,点击countine,待样品上完后,再将A1放入到平衡液中继续清洗柱子。 2),3)选择一种方式 4)洗脱:上样后用缓冲液尽量将穿透峰洗回基线。在manual里选择pump→gradient,按照自己的工艺选择targetB(100%)和length(10CV)。 5)设定收集:选择Frac→fractionation_900,输入每管收集体积,exectue。结束固定体积收集选择Frac→fractionation_stop_900,exectue。 6、清洗泵及卸下层析柱: 将A1和B1入口放入纯水中,启动pump wash purifier功能冲洗A泵和B泵及整个管路。然后再将A1和B1入口放入20%乙醇中,同样操作将乙醇冲满整个管路保存。系统给柱子一个慢流速,设置系统保护压力,然后先拆柱子的下端,正在滴水的时候将堵头拧上,再拆柱子的上端,最后拧上上端的堵头。整个过程防止气泡进入。 7、关闭电源: 从软件控制系统的第一个窗口unicorn manager点击退出,其他窗口不能单独关闭。 然后关闭AKTA主机电源,关闭电脑电源。