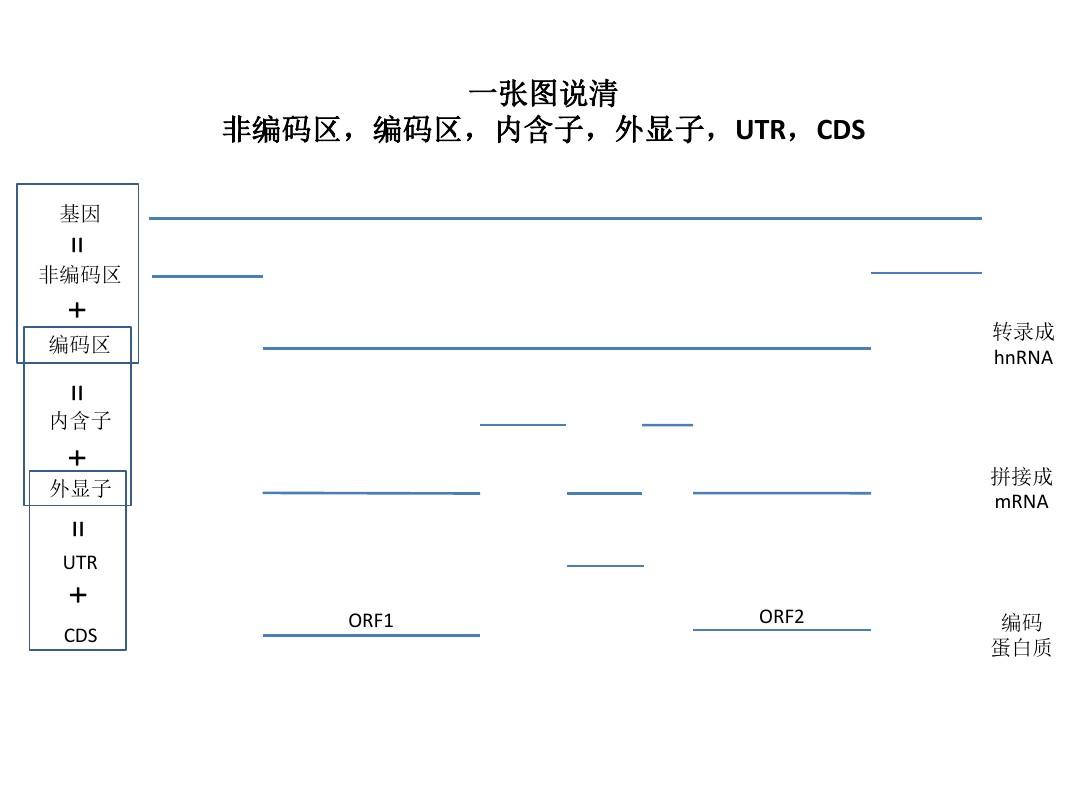
一张图说清非编码区,编码区,内含子,外显子,UTR,CDS
几种常见的基因测序技术的优缺点及应用复习过程
几种常见的基因测序技术的优缺点及应用
随着人类基因组计划的完成,人类对自身遗传信息的了解和掌握有了前所未有的进步。与此同时,分子水平的基因检测技术平台不断发展和完善,使得基因检测技术得到了迅猛发展,基因检测效率不断提高。从最初第一代以 Sanger 测序为代表的直接检测技术和以连锁分析为代表的间接测序技术,到 2005 年,以 Illumina 公司的 Solexa技术和 ABI 公司的 SOLiD 技术为标志的新一代测序(next-generation sequencing,NGS) 的相继出现,测序效率明显提升,时间明显缩短,费用明显降低,基因检测手段有了革命性的变化。其技术正向着大规模、工业化的方向发展,极大地提高了基因检测的检出率,并扩展了疾病在基因水平的研究范围。2009 年 3 月,约翰霍普金斯大学的研究人员在《Science》杂志上发表了通过 NGS外显子测序技术,发现了一个新的遗传性胰腺癌的致病基因PALB2,标志着 NGS 测序技术成功应用于致病基因的鉴定研究。同年,《Nature》发表了采用 NGS 技术发现罕见弗里曼谢尔登综合征MYH3 致病基因突变和《Nat Genet》发表了遗传疾病米勒综合征致病基因。此后,通过 NGS 技术,与遗传相关的致病基因不断被发现,NGS 技术已成为里程碑式的进步。2010 年,《Science》杂志将这一技术评选为当年“十大科学进展”。 近两年,基因检测成为临床诊断和科学研究的热点,得到了突飞猛进和日新月异的发展,越来越多的临床和科研成果不断涌现出来。同时,基因检测已经从单一的遗传疾病专业范畴扩展到复杂疾病和个体化应用更加广阔的领域,其临床检测范围包括高危疾病的新生儿筛查、遗传疾病的诊断和基因携带的检测以及基因药物检测用于指导个体化用药剂量、选择和药物反应等诸多方面的研究。目前,基因检测在临床诊断和医学研究的应用正越来越受到医生的普遍重视和引起研究人员的极大的兴趣。 本文介绍了几种 DNA 水平基因检测常见的方法,比较其优缺点和在临床诊断和科学研究中的应用,对指导研究生和临床医生课外学习,推进临床科研工作和提升科研教学水平有着指导意义。 1、第一代测序 1.1 Sanger 测序采用的是直接测序法。1977年,Frederick Sanger 等发明了双脱氧链末端终止法,这一技术随后成为最为常用的基因测序技术。2001 年,Allan Maxam 和 Walter Gibert 发明了 Sanger 测序法,并在此后的 10 年里成为基因检测的金标准。其基本原理即双脱氧核苷三磷酸(dideoxyribonucleoside triphosphate,ddNTP) 缺乏PCR 延伸所需的 3'-OH,因此每当 DNA 链加入分子 ddNTP,延伸便终止。每一次 DNA 测序是由 4个独立的反应组成,将模板、引物和 4 种含有不
41. ACMG全外显子测序指南.
ACMG全外显子测序指南 摘要:美国医学遗传学与基因组学学会(ACMG)以前为序列突变的解释提供了指导.1在过去十年中,随着高通量测序的出现,测序技术迅速发展。通过采用和利用下一代测序,临床实验室正在进行基因分型,单基因,基因组,外显子,基因组,转录组和遗传疾病表观遗传学检测的不断增加的遗传检测目录。由于复杂性增加,基因检测的这种转变伴随着序列解释的新挑战。在这方面,ACMG于2013年召集了一个由ACMG,分子病理学协会(AMP)和美国病理学家学会的代表组成的工作组,重新审视和修订了序列突变解释的标准和准则。该组由临床实验室主任和临床医生组成。本报告代表ACMG,AMP和美国病理学家利益相关者联盟组成的工作组的专家意见。这些建议主要适用于临床实验室使用的遗传检测的范围,包括基因分型,单基因,panel,外显子和基因组。本报告建议使用具体的标准术语- “致病性”,“可能致病性”,“不确定性意义”,“可能良性”和“良性”来描述在导致孟德尔病症的基因中鉴定的突变。此外,该建议描述了基于使用典型类型的突变证据(例如,群体数据,计算数据,功能数据,分离数据)的标准将突变分类为这五个类别的过程。由于本报告中描述的临床基因检测的分析和解释的复杂性增加,ACMG强烈建议临床分子遗传学检测应在经过临床实验室改进修订批准的实验室进行,结果由相关职业认证的临床分子遗传学家或分子遗传病理学家或同等学科专家进行解释。 关键词:ACMG实验室指导; 临床遗传检测; 解释;报告; 序列变异术语;突变报告 前言 临床分子实验室正在不断增加检测的新的序列突变,因为在检测患者标本时不断发现大量与基因疾病相关的基因。虽然一些表型与单个基因相关,但许多与多个基因相关。我们对任何给定序列突变的临床意义的理解是循序渐进的,其范围从那些几乎肯定是疾病致病性突变到几乎肯定是良性的突变。虽然以前的美国医学遗传学和基因组学会(ACMG)的建议提供了序列突变的解释类别和解释算法,但是这些建议没有提供定义的术语或详细的突变分类指南.1。本报告描述了
人全外显子组序列捕获及第二代测序
人全外显子组序列捕获及第二代测序 概述 外显子组是指全部外显子区域的集合,该区域包含合成蛋白质所需要的重要信息,涵盖了与个体表型相关的大部分功能性变异。外显子组序列捕获及第二代测序是一种新型的基因组分析技术:外显子序列捕获芯片(或溶液)可在同一张芯片上以高特异性和高覆盖率捕获研究者感兴趣的目标外显子区域,后续利用Solexa/SOLiD/Roche 454测序直接解析数据。 与全基因组重测序相比,外显子组测序只需针对外显子区域的DNA 即可,覆盖度更深、数据准确性更高,更加简便、经济、高效。可用于寻找复杂疾病(如:癌症、糖尿病、肥胖症等)的致病基因和易感基因等的研究。同时,基于大量的公共数据库提供的外显子数据,我们能够结合现有资源更好地解释我们的研究结果。 目前,SBC提供的外显子组序列捕获芯片是NimbleGen Sequence Capture 2.1M Human Exome Array及Agilent SureSelect Target Enrichment System(Human Exome)。 技术路线 以Nimblegen外显子捕获结合Solexa测序为例加以说明:基因组DNA首先被随机打断成500bp左右的片段,随后在DNA片段两端分别连接上接头。经过PCR库检合格后的DNA 片段与NimbleGen 2.1M Human Exome Array芯片进行杂交。去除未与芯片结合的背景DNA 后,将经过富集的外显子区域的DNA片段洗脱下来。这些DNA片段又随机连接成长DNA片段
后,再次被随机打断并在其两端加上测序接头,经过LM-PCR的线性扩增,在经qPCR质量检测合格后即可上机测序。 外显子组测序的实验流程示意图(www.nimblegen.com) 生物信息学分析流程图 研究内容 1.外显子组捕获与测序 将基因组DNA随机打断成片段,通过与人全外显子捕获芯片杂交富集外显子区域,通过第二代测序技术对捕获的序列进行测序。 2.基本数据分析 数据产出统计:对测序结果进行图像识别(Base calling),去除污染及接头序列;统计结果包括:测定的序列(Reads)长度、Reads数量、数据产量。 3. 高级数据分析 高级数据分析内容包括: (1)Clean reads序列与参考基因组序列比对; (2)目标外显子区域测序深度分析; (3)目标外显子区域一致序列组装;
基因测序技术的优缺点及应用
基因测序技术的优缺点及应用 随着人类基因组计划的完成,人类对自身遗传信息的了解和掌握有了前所未有的进步。与此同时,分子水平的基因检测技术平台不断发展和完善,使得基因检测技术得到了迅猛发展,基因检测效率不断提高。从最初第一代以 Sanger 测序为代表的直接检测技术和以连锁分析为代表的间接测序技术,到 2005 年,以Illumina 公司的 Solexa技术和 ABI 公司的 SOLiD 技术为标志的新一代测序 (next-generation sequencing,NGS) 的相继出现,测序效率明显提升,时间明显缩短,费用明显降低,基因检测手段有了革命性的变化。其技术正向着大规模、工业化的方向发展,极大地提高了基因检测的检出率,并扩展了疾病在基因水平的研究范围。2009 年 3 月,约翰霍普金斯大学的研究人员在《Science》杂志上发表了通过 NGS外显子测序技术,发现了一个新的遗传性胰腺癌的致病基因PALB2,标志着 NGS 测序技术成功应用于致病基因的鉴定研究。同年,《Nature》发表了采用 NGS 技术发现罕见弗里曼谢尔登综合征MYH3 致病基因突变和《Nat Genet》发表了遗传疾病米勒综合征致病基因。此后,通过 NGS 技术,与遗传相关的致病基因不断被发现,NGS 技术已成为里程碑式的进步。2010 年,《Science》杂志将这一技术评选为当年“十大科学进展”。 近两年,基因检测成为临床诊断和科学研究的热点,得到了突飞猛进和日新月异的发展,越来越多的临床和科研成果不断涌现出来。同时,基因检测已经从单一的遗传疾病专业范畴扩展到复杂疾病和个体化应用更加广阔的领域,其临床检测范围包括高危疾病的新生儿筛查、遗传疾病的诊断和基因携带的检测以及基因药物检测用于指导个体化用药剂量、选择和药物反应等诸多方面的研究。目前,基因检测在临床诊断和医学研究的应用正越来越受到医生的普遍重视和引起研究人员的极大的兴趣。 本文介绍了几种 DNA 水平基因检测常见的方法,比较其优缺点和在临床诊断和科学研究中的应用,对指导研究生和临床医生课外学习,推进临床科研工作和提升科研教学水平有着指导意义。 1、第一代测序 1.1 Sanger 测序采用的是直接测序法。1977年,Frederick Sanger 等发明了双脱氧链末端终止法,这一技术随后成为最为常用的基因测序技术。2001 年,Allan Maxam 和 Walter Gibert 发明了 Sanger 测序法,并在此后的 10 年里成为基因检测的金标准。其基本原理即双脱氧核苷三磷酸(dideoxyribonucleoside triphosphate,ddNTP) 缺乏PCR 延伸所需的 3'-OH,因此每当 DNA 链加入分子 ddNTP,延伸便终止。每一次 DNA 测序是由 4个独立的反应组成,将模板、引物和 4 种含有不同的放射性同位素标记的核苷酸的ddNTP 分别与DNA 聚合酶混合形成长短不一的片段,大量起始点相同、终止点不同的 DNA 片段存在于反应体系中,具有单个碱基差别的 DNA 序列可以被聚丙烯酰胺变性凝胶电泳分离出来,得到放射性同位素自显影条带。依据电泳条带读取DNA 双链的碱基序列。 人类基因组的测序正是基于该技术完成的。Sanger 测序这种直接测序方法具有高度的准确性和简单、快捷等特点。目前,依然对于一些临床上小样本遗传疾病基因的鉴定具有很高的实用价值。例如,临床上采用 Sanger 直接测序 FGFR 2 基因证实单基因 Apert 综合征和直接测序 TCOF1 基因可以检出多达 90% 的
50个全外显子测序揭示人类的高原适应机制
50个全外显子测序揭示人类的高原适应机制 Xin Yi等。 Science 329, 75 (2010); DOI: 10.1126/science.1190371 50个全外显子测序揭示人类的高原适应机制 生活于青藏高原的藏族人表现出了对极端高原环境的遗传适应性。我们对50个藏族人进行全外显子基因组测序,它们包含了92%的人类基因编码序列,人均覆盖度为18倍。基因分析显示了该特殊人群等位基因频率的变化,表明这些人对高原环境具有很强的适应性。 研究显示,表现出最强自然选择信号的基因是编码内皮细胞含PAS结构域蛋白-1(EPAS1)的基因,这是一个参与应答缺氧的转录因子。研究显示,EPAS1基因的一个单核苷酸多态性(SNP)在78%的藏族和汉族人群中存在差异,这是目前发现的速率改变最快的等位基因。该单核苷酸多态性与红细胞丰度的关联分析也支持EPAS1改变在适应缺氧环境中的作用,进一步表明它是适应高原环境的一个重要的遗传位点。 在广袤的大自然中生存的人类可能会存在文化和基因上的适应。其中人类面临的最严厉的环境挑战就是高海拔地区(如青藏高原)的低含氧量。这一地区的许多居民在海拔4000米以上居住,那里的氧气浓度比海平面大约低40%。藏族对缺氧环境有着他们自己的遗传适应性,如出生体重(1),血红蛋白水平(2),婴儿(3)和运动后的成年人(4)血液中的氧饱和度。这些结果暗示了高原适应机制的自然选择历史,我们对整个基因组的遗传差异进行分析,可能会发现这一点。 我们对中国西藏自治区海拔4300米以上(5)的两个村庄里的50个非亲个体进行全外显子基因组测序。针对将近两万个基因的外显子和侧翼区的34Mb序列,利用罗氏NimbleGen公司(威斯康星州麦迪逊市)的2.1M外显子序列捕获芯片(6)将其富集。测序采用了Illumina公司(加利福尼亚州圣地亚哥市)的基因组分析仪II平台,并使用序列比对程序SOAP(7)将测序片段比对到人类参考基因组序列上[美国生物技术信息中心(NCBI) 36. 3版]。 1深圳华大基因研究院,中国深圳,518083。 2中国科学院研究生院,中国北京,100062。 3加州大学伯克利分校综合生物学与统计系,美国加州,94820。4华南理工大学生物系本科创新班,中国广州,510641。 5西藏自治区人民医院,中国拉萨,850000。 6加州大学戴维斯分校进化与生态学系,美国加州,95616。 7哥本哈根大学生物系,丹麦哥本哈根,1165。 8华南理工大学理学院本科创新班,中国广州,510641。 9深圳大学医学院基因组研究所,中国深圳,518060。 10拉萨市人民医院,中国拉萨,850000。 11西藏军区总医院,中国拉萨,850007。 12西双版纳傣族自治州人民医院,中国云南景洪,666100。 *以上机构及相关人员对本研究作出了贡献。如有疑问请联系: E-mail:wangjian@genomics.org.cn ( Ji.W.); wangj@genomics.org.cn ( Ju.W.); rasmus_nielsen@berkeley.edu (R.N.) 全外显子测序的平均深度为18倍(表S1),但这并不能保证个别基因型的准确性。因此,我们用贝叶斯统计法(5)估算出每个可能的基因型概率,从而估算出单核苷酸多态性(SNP)的概率和每个位点的人类等位基因频率。在藏族样本中总共151825个SNPs有超过50%被识别出是可变的,有101668个超过99%的SNP是可变的(表S2)。Sanger测序验证了56个SNPs 中的53个,至少包含95%的SNP和3%~50%的次等位基因频率。等位基因频率的估算值显示存在过量的低频变异(图S1),特别是在非同义SNPs中。 该数据与来自于北京的40个汉族人的基因组进行比较[样本来自于中测检测(CHB)人类基因组单体型图计划(HapMap),属于1000个基因组计划的一部分(http://1000genomes.org)],测序得出汉族人均大约4倍的覆盖度。北京的海拔不超过50米,几乎所有的汉族人来自于海拔2000米以下。在较低的遗传分化基础上汉族人样本与藏族人样本形成鲜明的对比(F ST= 0.026)。这两个藏族村庄是体现该族遗传结构的最小单位(F ST= 0.014),因此,我们可以将他们当着整个群体进行分析。我们观察到,汉藏之间的等位基因频率有着很强的协方差(图1),但是过量的SNPs在汉族中频率很低,在藏族中频率中等。 从两个群体同义位点的二维频谱,可以估算出人类历史模型(8)。最佳拟合模型表明,藏族和汉族人群在2750年前出现分化,汉族人口从最初的小规模逐渐增大,藏族人口从最初的大规模逐渐减少(图S2)。这估计是由于藏族人移民至汉族区,双方长期相互渗透所造成的。
外显子捕获结题报告
外显子捕获结题报告2010-11-22
内容 1 项目信息 (1) 2 工作流程介绍 (2) 2.1 Agilent液相捕获平台 (2) 2.2 NimbleGen 液相捕获平台 (3) 2.3 生物信息分析流程 (4) 3 分析报告 (5) 结果 (5) 3.1 标准生物信息分析 (5) 3.1.1 数据产出统计 (5) 3.1.2 目标区域单碱基深度分布图 (6) 3.1.3外显子捕获测序的均一性 (7) 3.1.4一致序列组装和SNP检测 (7) 3.1.5 SNP注释 (8) 3.1.6插入/缺失(indels)检测 (9) 3.1.7插入/缺失(indels)注释 (9) 3.2个性化分析 (9) 3.2.1氨基酸替换预测 (9) 3.2.2群体SNP检测和等位基因频率估计 (12) 3.2.3孟德尔遗传病分析 (13) 3.2.4 NGS-GW AS 分析 (14) 3.2.5正向选择信号的检测 (14) 4 数据分析方法说明 (15) 4.1信息分析软件及常用参数介绍 (15) 4.2参考数据库 (16) 4.3数据文件格式 (17)
1 项目信息 PROJECT NAME CONTRACT NUMBER SAMPLE INFORMATION Species Information Genome Information Additional Information CUSTOMER INFORMATION PI Contact Person Company Name Contact Methods Name Tel E-mail Name Tel E-mail CONTACT INFORMATION (BGI) Sales Information Name Tel E-mail Name Tel E-mail Customer Service Name Tel E-mail Name Tel E-mail PROJECT DIRECTOR APPROVAL THE RESULTS HAVE BEEN APPROVED AND CAN BE SUBMITTED Signature: Date:
几种常见的基因测序技术的优缺点及应用
几种常见的基因测序技术的优缺点及应用 发布时间:2014-07-19 来源:毕业论文网 随着人类基因组计划的完成,人类对自身遗传信息的了解和掌握有了前所未有的进步。与此同时,分子水平的基因检测技术平台不断发展和完善,使得基因检测技术得到了迅猛发展,基因检测效率不断提高。从最初第一代以Sanger 测序为代表的直接检测技术和以连锁分析为代表的间接测序技术,到2005 年,以Illumina 公司的Solexa技术和ABI 公司的SOLiD 技术为标志的新一代测序(next-generation sequencing,NGS) 的相继出现,测序效率明显提升,时间明显缩短,费用明显降低,基因检测手段有了革命性的变化。其技术正向着大规模、工业化的方向发展,极大地提高了基因检测的检出率,并扩展了疾病在基因水平的研究范围。2009 年 3 月,约翰霍普金斯大学的研究人员在《Science》杂志上发表了通过NGS外显子测序技术,发现了一个新的遗传性胰腺癌的致病基因PALB2,标志着NGS 测序技术成功应用于致病基因的鉴定研究。同年,《Nature》发表了采用NGS 技术发现罕见弗里曼谢尔登综合征MYH3 致病基因突变和《Nat Genet》发表了遗传疾病米勒综合征致病基因。此后,通过NGS 技术,与遗传相关的致病基因不断被发现,NGS 技术已成为里程碑式的进步。2010 年,《Science》杂志将这一技术评选为当年“十大科学进展”。 近两年,基因检测成为临床诊断和科学研究的热点,得到了突飞猛进和日新月异的发展,越来越多的临床和科研成果不断涌现出来。同时,基因检测已经从单一的遗传疾病专业范畴扩展到复杂疾病和个体化应用更加广阔的领域,其临床检测范围包括高危疾病的新生儿筛查、遗传疾病的诊断和基因携带的检测以及基因药物检测用于指导个体化用药剂量、选择和药物反应等诸多方面的研究。目前,基因检测在临床诊断和医学研究的应用正越来越受到医生的普遍重视和引起研究人员的极大的兴趣。 本文介绍了几种DNA 水平基因检测常见的方法,比较其优缺点和在临床诊断和科学研究中的应用,对指导研究生和临床医生课外学习,推进临床科研工作和提升科研教学水平有着指导意义。 1、第一代测序 1.1 Sanger 测序采用的是直接测序法。1977年,Frederick Sanger 等发明了双脱氧链末端终止法,这一技术随后成为最为常用的基因测序技术。2001 年,Allan Maxam 和Walter Gibert 发明了Sanger 测序法,并在此后的10 年里成为基因检测的金标准。其基本原理即双脱氧核苷三磷酸(dideoxyribonucleoside triphosphate,ddNTP) 缺乏PCR 延伸所需的 3'-OH,因此每当DNA 链加入分子ddNTP,延伸便终止。每一次DNA 测序是由4个独立的反应组成,将模板、引物和 4 种含有不同的放射性同位素标记的核苷酸的ddNTP 分别与DNA 聚合酶混合形成长短不一的片段,大量起始点相同、终止点不同的DNA 片段存在于反应体系中,具有单个碱基差别的DNA 序列可以被聚丙烯酰胺变性凝胶电泳分离出来,得到放射性同位素自显影条带。依据电泳条带读取DNA 双链的碱基序列。 人类基因组的测序正是基于该技术完成的。Sanger 测序这种直接测序方法具有高度的准确性和简单、快捷等特点。目前,依然对于一些临床上小样本遗传疾病基因的鉴定具有很高的实用价值。例如,临床上采用Sanger 直接测序FGFR 2 基因证实单基因Apert 综合征和直接测序TCOF1 基因可以检出多达90% 的与Treacher Collins 综合征相关的突变。值得注意的是,Sanger 测序是针对已知致病基因的突变位点设计引物,进行PCR 直接扩增测序。
全外显子组检测技术参数要求
附件:全外显子组检测技术参数要求 一、公司资质: 1.拥有先进的高通量二代测序平台和高性能计算平台; 2.具有短期处理大量样本,进行全外显子组和全基因组测序的经验; 3.实验室具有国内或国外权威机构的资质认证; 4.*应标的公司必须通过医学遗传中心选送的样本测试(三个以上生物学重复),并且需交 付原始下机数据,以中心提供的标准化流程统一进行质量评估。 二、技术参数: 1)污染防控 具有独立的实验方法进行样本身份鉴定,可追溯样本间发生的错误 2)测序质量 1.Q20平均比例在90%以上。 2.Q30平均比例在85%以上。 3.GC content 分布无明显偏移。 3)测序深度、覆盖度统计 下文涉及的数据均为经过去接头、比对、排序和去重后的有效数据。数据统计涉及的相关软件除特别说明外,应使用默认参数。 1.数据质量要求: 1)Mapped unique reads相对总reads的比例(PCT_PF_UQ_READS_ALIGNED)不得低于
99% 2)有效数据总量(PF_UQ_BASES_ALIGNED)不得低于10G 3)On targeted bases相对总bases的比例(PCT_USABLE_BASES_ON_BAIT)不得低于50% 4)On and near targeted bases相对总bases的比例(PCT_SELECTED_BASES)不得低于 80% 5)全外显子碱基10X覆盖率(PCT_TARGET_BASES_10X)不得低于95% 6)全外显子碱基30X覆盖率(PCT_TARGET_BASES_30X)不得低于80% 7)全外显子组各区域覆盖的一致性统计要求:80%以上的target region的normalized coverage值不得低于0.3 三、项目内容: 500例耳聋患者全外显子组测序技术服务
06-重测序、外显子组试卷答案.pdf
一、名词解释 1.比对将测序序列比对到参考基因组序列 2.单核苷酸多态性主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多 态性 3.三体家系样本,父亲,母亲,和孩子家系外显子组中的一组家系样本 4.小的核苷酸的插入缺失 5.拷贝数变异是由基因组发生重排而导致的,一般指长度为1 kb以上的基因组大片段 的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复 二、填空题 1.bwa GATK 2.1% 3. 全基因组重测序全外显子组区域捕获测序 4. snp indel CNV SV 5. 5 50X 6. 1.5 3.5 7.0.1% 1% 8.液氮冻存-80冰箱冻存 9.血液 10.Agilent SureSelect All Exon V4 (+ UTR), NimbleGen SeqCap EZ Human Exome Library v3.0, Illumina TruSeq Exome Enrichment Kit 51M(71M), 64M, 62M 三、问答题 1. 建库:将基因组DNA经Covaris破碎仪随机打断成长度为180-280bp的片段,末端修复和加A 尾后在片段两端分别连接上接头制备 DNA 文库。 捕获:带有特异index的文库pooling后与多达543,872个生物素标记的探针进行液相杂交,再使用带链霉素的磁珠将20,965个基因的334,378个外显子捕获下来。 扩增及测序:经PCR线性扩增后进行文库质检,合格即可进行测序。 2. 人类85%的疾病位点位于编码区 域外显子组可以提供更深的测序 深度外显子组花费更低 3. 数据质控,比对到参考基因组,去重复重校正,预测个体snp和indel,预测体细胞突变,预测CNV和SV,候选位点注释 4. 常染色体显性遗传 常染色体隐形遗传 伴X 染色体显性遗传 伴X 染色体隐性遗传 伴Y 染色体遗传 找Denovo mutation,只在患病孩子有,不在健康父母里存在的位点 5.
寻找基因外显子、内含子的几种方法
寻找基因外显子、内含子的几种方法 以人类的wnt3a基因为例 一、http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=gene 1、进入ncbi的gene数据库【网址: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=gene】 2、在for栏输入wnt3a,点击limits 3、在All fields 栏选择Gene Name,在Homo sapiens前打勾,点击go 4、出现下图,点击wnt3a 5、鼠标左键点击NC-000001.9,选择Genbank
或在Genomic栏下点击Genbank【图中圈出的部分】 6、出现下图,图中画线部分就是外显子的位点【注意不是图中圆圈的部分】外显子分别为1~149 15617~15858 43606~43871 51936~54210
二、http://www.genome.ucsc.edu/ 1、进入http://www.genome.ucsc.edu/的网页 2、点击Gene Sorter 3、在genome栏输入human,在search栏输入wnt3a,点击go
4、出现下图,点击图中圈出来的部分: 5、出现下图,点击sequence 6、出现下图,点击Genomic 7、出现下图,点击submit
8、出现序列,其中外显子用大写字母,内含字用小写字母。 9、将其拷贝到word中,鼠标定位到大小写接头的位点,进行定位统计。 疑问:奇怪的是得出的结果与方法一、方法三不符,不知是什么原因?请大家指点,看看是哪里错了。 这种方法的介绍见:http://www.dxy.cn/bbs/actions/archive/post/6145797_1.html 三、http://www.ensembl.org/
人外显子测序
人外显子测序 药明康德基因中心,陆桂1. 什么是外显子测序(whole exon sequencing)? 外显子组测序是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。外显子测序相对于基因组重测序成本较低,对研究基因的SNP、Indel 等具有较大的优势,但无法研究基因组结构变异如染色体断裂重组等。 2. 外显子捕获试剂盒有哪些? 目前主要有Roche、Illumina和Agilent三家的外显子捕获试剂。Nimblegen和Illumina的捕获试剂盒中的探针是DNA探针,化学性质稳;Agilent的捕获试剂盒是RNA探针,有可能RNA 不是很稳定。 3. 外显子捕获效率是什么? 外显子测序过程中要用到杂交过程。在人的染色体上有许多与外显子有同源性的部分,这些有同源性的部分很可能在杂交过程中也被捕获下来。所以,测到的序列中,有一部分不是外显子序列。我们把测序得是外显子的部分占全部测序序列的比列称为捕获效率。 Nimblegen大约是70% Agilent大约是60% Illumina大约是50% 4. 外显子测序一般建议做多少倍的覆盖? 一般做100X或者150X。较高的覆盖倍数,对于测异质性的遗传变质,可以发现小比例的突变。另外,外显子测序的覆盖不是很均匀,这样较高的平均覆盖率有利于保证大部分的区域有足够的覆盖倍数。 5. 外显子测序能够测出多大的片段缺失? 大致能测出50bp的片段缺失。目前的测序主要还是用Hiseq 2000,单侧的测长就是100bp。由于外显子测序的覆盖很不平均,所以如果有大段的缺失,无法判断是因为杂交没有捕获到,还是因为缺失。目前能够测到的,就是在一个read中发现的缺失。一个read的长度也就是100bp,所以大到50bp以下的片段缺失可以从外显子测序中测出来。 6. 外显子捕获可以做CNV吗? 外显子测序因为有一个杂交捕获的过程,这样就会有一个杂交捕获效率的问题。各个外显子的杂交效率是不同的,其同源竞争的情况也不同,所以不同的外显子的覆盖率的差异就很大。所以一般情况下,外显子测序不能用于CNV的检测。但在癌症研究中,利用癌组织和癌旁组织对照,可以检测CNV。 现在我们有另外两种常规方法来检测CNV,一种是全基因组重测序,另外一种是用Affymetrix SNP6.0的芯片来测。其中Affymetrix SNP6.0的检测费用大约只有全基因测序费用的1/10,是一个相对经济的手段。 7. 外显子测序的优点是什么?
华大智造外显子捕获测序解决方案
华大智造外显子捕获测序解决方案 概述 随着测序技术发展和成本降低,临床外显子组测序(cWES)和全外显子测序(WES)在遗传病检测领域崭露头角。外显子测序借助捕获探针(DNA或RNA)对人基因组约1-2%的区域测序,可覆盖绝大多数基因的编码序列和>99%(临床基因组资源库,ClinGen)疾病相关区域。华大智造基于自有的探针合成平台和高通量测序仪(MGISEQ/BGISEQ 系列),能为客户提供外显子测序一站式解决方案。 图1 外显子测序示意图(以MGI测序平台为例)
MGIEasy 外显子组捕获V5探针试剂套装 MGIEasy 外显子组捕获V5探针试剂套装除了涵盖传统外显子探针覆盖的区域,还有针对性的做了探针优化,保证了生育健康、新生儿、心脑血管、遗传性肿瘤、单基因病、安全用药、个人基因组、遗传性耳聋、免疫缺陷、线粒体缺陷等致病基因的全覆盖。 产品亮点 ●探针区域69Mb ●更多的疾病致病位点 ●更优的数据利用率 ●稳定而高效的捕获效率 技术优势 数据库覆盖情况 MGI V5与竞品(Vendor A6/N3/I)比,有更多的独有区域,涵盖了华大自主研发的 图2 CCDS、GENCODE、UCSC、miRBase和RefSeq数据库基因数量覆盖情况 基因覆盖更全面
MGI V5能100%覆盖的基因数达到455个,远高于A5 (125个)、N3 (33个)和I (357个),其独有100%覆盖基因数达到160个,是A5和N3之和。 BBS10基因是巴比二氏综合征的致病基因,MGI V5完整涵盖了基因区和内含子区,其中包括ClinVar数据库中报道的已知临床突变位点。 基因覆盖均一性更优 MGI V5在测序深度达到100x时,96%的区域覆盖度均能达到20X以上。与竞品N3和I共有的区域,MGI V5显示了更优秀的覆盖均一性。 性能比较 图3 100%覆盖的基因数和BBS10基因覆盖情况 图4 >96%区域达到20X覆盖图5 共有区域的覆盖更均一
外显子组测序
技术参数 样品要求捕获平台测序策略 测序深度 项目周期 外显子组测序 37天 1. 单基因病/复杂疾病有效测序深度50X以上 2. 肿瘤有效测序深度100X以上 注:可根据老师研究目的进行更高深度测序 HiSeq PE150 Agilent SureselectXT Custom Kit 样品总量:≥1.0 μg DNA (提取自新鲜及冻存样本) ≥1.5 μg DNA (提取自FFPE样本)样品浓度:≥20 ng/μl 参考文献 外显子组测序(Whole Exome Sequencing,WES)是利用探针杂交富集外显子区域的DNA序列,通过高通量测序,发现与蛋白质功能变异相关遗传突 变的技术手段。相比于全基因组测序,外显子组测序更加经济、高效。 1. 直接对蛋白编码序列进行测序,找出影响蛋白结构的变异 2. 高深度测序,可发现常见变异及频率低于1%的罕见变异 3. 针对外显子组区域测序,约占基因组的1%,有效降低费用,周期和工作量 技术优势 生物信息分析 基本信息分析 1. 数据质控:去除接头污染和低质量数据 2. 与参考序列进行比对、统计测序深度及覆盖度 3. SNP/InDel检测、注释及统计 4. Somatic SNV/InDel检测、注释及统计(成对样本) 高级信息分析(单基因病) 高级信息分析(复杂疾病) 高级信息分析(癌症) 1. 突变位点过滤 2. 显/隐性遗传模式分析(需老师提供家系信息) 2.1. 显性遗传模式分析 2.2. 隐性遗传模式分析 3. 候选基因功能注释 4. 新生突变筛选及分析(成三/成四家系) 4.1. de novo mutation 筛选 4.2. 新生突变速率计算 5. 候选基因功能富集 6. 蛋白互作网络分析(PPI) 7. 基因显著性分析 (推荐20对Case/Control or trios样本) 1. 突变位点过滤 2. 显/隐性遗传模式分析(需老师提供家系信息) 2.1. 显性遗传模式分析 2.2. 隐性遗传模式分析 3. 候选基因功能注释 4. 基因功能及通路分析 5. 家系连锁分析 6. 纯合子区域(ROH)分析 1. 易感基因筛查 2. NMF突变特征及突变频谱分析 3. 已知驱动基因筛选 4. 高频突变基因统计及通路富集分析 5. MRT高频突变基因相关性分析 6. OncodriveCLUST驱动基因预测 7. 高频CNV分布及重现性分析 8. 肿瘤纯度/倍性分析 9. 异质性/克隆结构分析 10. NovoDrug高频突变基因靶向用药预测11. NovoDR耐药突变筛选12. 基因组变异Circos图展示 案例解析 [案例一] 单基因病研究:外显子测序解析卵巢早衰的遗传因素[12] 卵巢早衰通常是指女性40岁之前闭经,1%的妇女患有此病,病因复杂,被认为受到遗传因素的影响。这项研究利用外显子测序技术首次在中东家系1(MO1DA)的卵巢早衰病人中发现了减数分裂基因中的STAG3基因突变可以导致隐性遗传卵巢早衰,也在小鼠动物模型和卵巢早衰病患中得到了证实。为探索卵巢早衰或卵巢功能不全的发生机理,以及阐明该病的临床高度异质性和遗传病因复杂性开辟了一个新的研究途径。 [案例二] 复杂疾病研究:外显子测序鉴定肌萎缩性脊髓侧索硬化症(ALS)的致病 基因[13] 肌萎缩性脊髓侧索硬化症(ALS),又称为渐冻症,是一种成年型的神经退行性疾病。本研究选取了47个父母+患病儿的ALS家系,利用全外显子测序寻找De novo mutatio n 。发现了25个de novo突变基因,进行功能聚类分析,锁定了1个与染色质包装、神经树突生长相关的基因CREST,后期通过细胞试验验证了该基因突变会影响神经元的伸展,证实CREST突变与ALS相关。 [案例三] 癌症研究:外显子测序研究局限性肺腺癌瘤内异质性[14] 本研究采用多区域取样分析瘤内异质性的研究思路,对11位患者的局限性肺腺癌的48个肿瘤样品进行了外显子测序。共鉴定出7269个体突变,其中21个是已知的与癌症相关的基因突变,76% 的体突变及21个已知癌症基因突变中的20个都可以在同一肿瘤的所有区域样品中检测到,表明对肿瘤的某一区域进行单次活检,以适当的深度对其测序,可以鉴别出绝大多数突变。而前期关于肾透明细胞癌的研究结果表明,肿瘤不同区域样品的共有突变仅占突变总数的31%~37%,说明肿瘤异质性在不同癌种间存在差异。 [1] Krawitz PM, Schweiger MR, R?delsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome[J]. Nature Genetics, 2010, 42(10): 827-829.[2] Liu Y, Gao M, Lv YM, et al. Confirmation by exome sequencing of the pathogenic role of NCSTN mutations in acne inversa (hidradenitis suppurativa) [J]. Journal of Investigative Dermatology,2011, 131(7): 1570-1572. [3] Wei A H, Zang D J, Zhang Z, et al. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism[J]. Journal of Investigative Dermatology, 2013, 133(7): 1834-1840. [4] Sanna-Cherchi S, Sampogna R V, Papeta N, et al. Mutations in DSTYK and dominant urinary tract malformations[J]. New England Journal of Medicine, 2013, 369(7): 621-629.[5] Musunuru K, Pirruccello J P , Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia[J]. New England Journal of Medicine, 2010, 363(23): 2220-2227. [6] O'Roak B J, Deriziotis P , Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations[J]. Nature genetics, 2011, 43(6): 585-589. [7] Jones S, Wang T L, Shih I M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma[J]. Science, 2010, 330(6001): 228-231. [8] Yan X J, Xu J, Gu Z H, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia[J]. Nature Genetics, 2011, 43(4): 309-315. [9] Rudin C M, Durinck S, Stawiski E W, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer[J]. Nature Genetics, 2012, 44(10): 1111-1116. [10] Yi X, Liang Y, Huerta-Sanchez E, et al. Sequencing of 50 human exomes reveals adaptation to high altitude[J]. Science, 2010, 329(5987): 75-78. [11] Tennessen J A, Bigham A W, O’Connor T D, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes[J]. Science, 2012, 337(6090): 64-69. [12] Caburet S, Arboleda V A, Llano E, et al. Mutant cohesin in premature ovarian failure[J]. New England Journal of Medicine, 2014, 370(10): 943-949.[13] Chesi A, Staahl B T, Jovicic A, et al. Exome sequencing to identify de novo mutations in sporadic ALS trios[J]. Nature Neuroscience, 2013, 16(7): 851-855.[14] Zhang J, Fujimoto J, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multi region sequencing[J]. Science, 2014, 346: 256-259. 群体研究 藏族人高原适应性研究[10];深度解析人类罕见遗传变异[11];…… 图1 STAG3 基因结构图 (红色箭头为 STAG3 基因突变位置) 图2 ALS家系图及CREST突变功能验证 图3 产生化疗抗性的个体样本中体突变的数量及频率
基因捕获
什么是基因陷阱或基因捕获 (gene trap)? 基因陷阱或基因捕获(gene trap)是通过在基因组中创造随机插入突变,来直接获得分子特征。基因陷阱或基因捕获载体包含一个无启动子的报告基因或选择标记,它能在插入位置(内含子)激活所在基因表达。因这系列方法酷似以报道基因为诱饵来捕获基因,故得名基因陷阱或基因捕获。换言之,它主要依靠报告基因的随机插入来产生融合转录物或融合蛋白,通过检测报道基因而推知基因及其功能。一般常用的报道基因有GUS、绿色荧光蛋白(GFP)、Lc基因。 在此基础上,还发展了启动子陷阱或启动子捕获(promoter trap)与增强子陷阱或增强子捕获(enhancer trap)。启动子陷阱或启动子捕获是通过将报道基因插入到细胞基因组的外显子上,如果发现它与细胞基因组基因被共同转录或表达,则可推知该报道基因附近有启动子。增强子陷阱或增强子捕获是将某报道基因与一个精巧的启动子相连,组成增强子陷阱重组体,它不会自主起始转录,需要由被插入的细胞基因组中的增强子帮助才可转录。若报道基因得以表达,则可推知插入位点附近有增强子或有基因。 图1:在被“捕获”基因的启动子的转录控制下,报告基因与插入位置的内源基因整合。融合的转录体由上游外显子和报告基因组成。在载体中,多聚腺苷酸信号限制到内源转录单位的最后一个外显子。通常采
用外显子陷阱和内含了陷阱两类。内含子陷阱包括一个剪接接受子序列(splice acceptor,SA)(在无启动子报告基因最上游)。外显子陷阱没有剪接接受子序列,在插入外显子后激活报告基因表达。(Figure 1.Integration within an endogenous gene places the reporter gene under the transcriptional control of the "trapped" gene's promoter. A fusion transcript is generated between upstream exons and the reporter gene. The polyadenylation signal (pA) within the vector defines the final exon of the endogenous transcription unit. Two types of vectors are commonly used, each of which can be introduced by electroporation or retroviral infection. The "intron trap" includes a splice acceptor sequence immediately upstream of a promoterless reporter gene that is activated following insertions in introns of genes. The "exon trap" lacks a splice acceptor and is designed to activate the reporter following insertions in exons.) 更多的信息参阅国际基因陷阱或基因捕获联合会(IGTC, International Gene-Trap Consortium)网站:http://www.igtc.ca/FAQ.html 基因陷阱或基因捕获有什么特点、优 势和劣势? 基因陷阱和启动子陷阱都有位置限制。基因陷阱重组体由报道基因和剪接接受子或部位(splice acceptor,SA)组成(接受体剪接部位在报道基因上游),该重组体需要插入到细胞基因组的内含子中随着基因转录和表达。如能检测到融合转录物或融合蛋白,就可证明插入位置附近有基因存在。启动子陷阱或启动子捕获需插入到内含子。因为增强子的作用特点,其位置与基因的位置可近可远,所以增强子陷阱不易定位基因。另外,对启动子陷阱和基因陷阱而言,插入可能导致基因失活。基因陷阱的优势在于它只在表达水平上定位基因,细胞基因本身的转录和