JJF气相色谱仪质谱联用仪

台式气相色谱-质谱联用仪校准规范
1 范围
本规范适用于离子阱和四极杆型台式气相色谱-质谱联用仪(以下简称台式GC-MS)的校准,其它类型台式GC-MS 的校准可参照此规范进行。
2 引用文献
JJF 1001―1998 通用计量术语及定义
JJF 1059―1999 测量不确定度评定与表示
GB/T 15481―1995 校准和检验实验室能力的通用要求
GB/T 6041―2002 质谱分析方法通则
JJG (教委) 003―1996 有机质谱仪检定规程
JJG 700―1999 气相色谱仪检定规程
OIML/TC16/SC2/R83 Gas chromatograph/mass spectrometer system for analysis of rganic pollutants in water
使用本规范时,应注意使用上述引用文献的现行有效版本。
3 术语和计量单位
分辨力(resolution)
分辨两个相邻质谱峰的能力,对于台式GC-MS以某离子峰峰高50%处的峰宽度(简称半峰宽)表示,记为W1/2,单位 u。
基线噪声(baseline noise)
基线峰底与峰谷之间的宽度,单位计数。
信噪比(signal-to-noise ratio)
待测样品信号强度与基线噪声的比值,记为S/N。
质量色谱图(mass chromatogram)质谱仪(和色谱图是两回事)质谱仪在一定质量范围内自动重复扫描所获得的质谱数据,可以不同形式再现,其中以一个或多个离子强度随时间变化的谱图,称为质量色谱图。
质量准确性(mass accuracy)
仪器测量值对理论值的偏差。
u (atomic mass unit)
原子质量单位。
4 概述
气相色谱-质谱联用仪是将气相色谱仪与质谱仪通过一定接口耦合到一起的分析仪器。样品通过气相色谱的分离后的各个组分依次进入质谱检测器,组分在离子源被电离,产生带有一定电荷、质量数不同的离子。不同离子在电场和/或磁场中的运动行为不同,采用不同质量分析器把带电离子按质荷比(m/z)分开,得到依质量顺序排列的质谱图。通过对质谱图的分析处理,可以得到样品的定性、定量结果。气相色谱-质谱联用仪主要包括气相色谱系统(一般不带检测器)、离子源、质量分析器、检测器、真空系统和计算机系统等几部分。
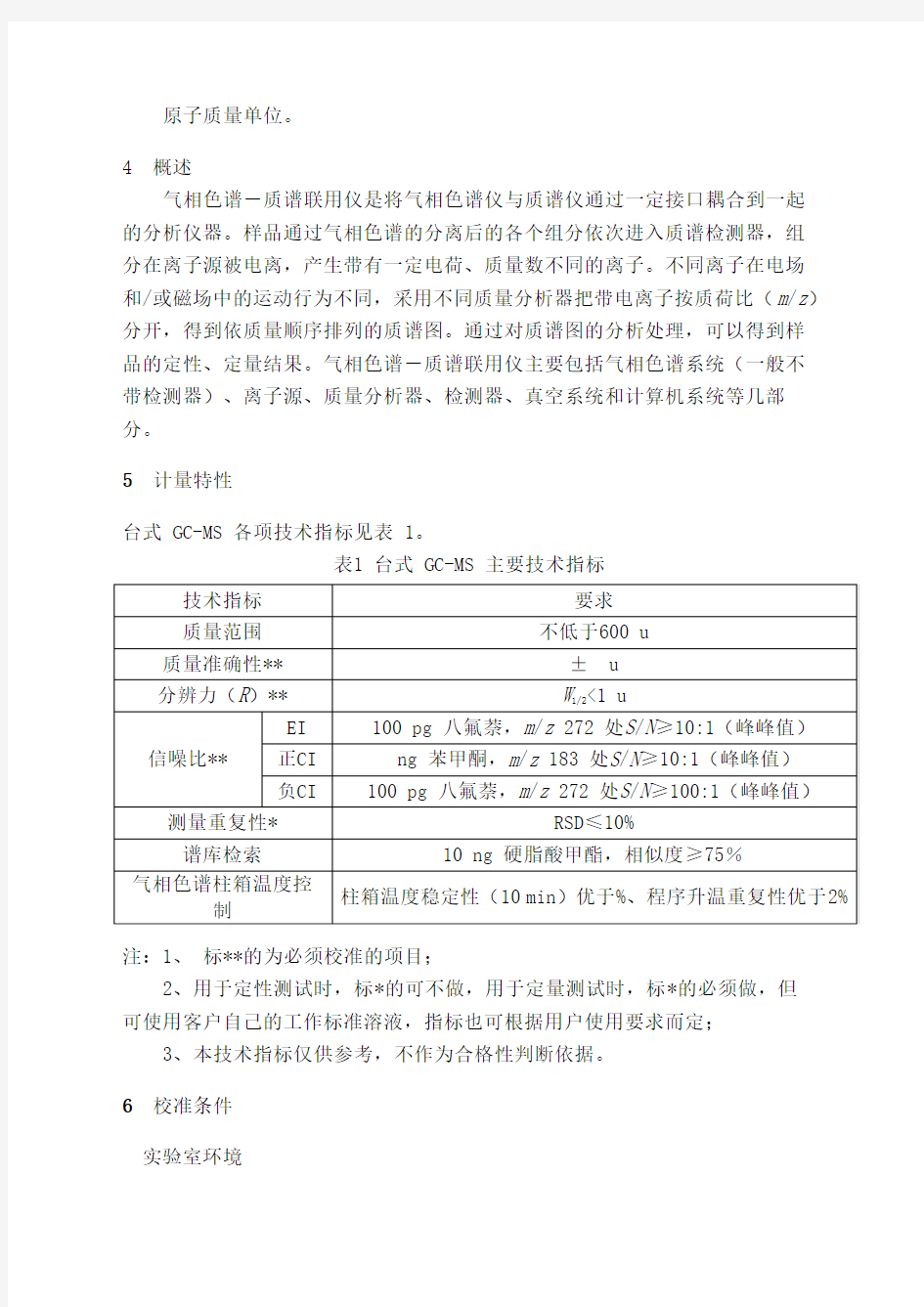
5 计量特性
台式 GC-MS 各项技术指标见表 1。
表1 台式 GC-MS 主要技术指标
注:1、标**的为必须校准的项目;
2、用于定性测试时,标*的可不做,用于定量测试时,标*的必须做,但
可使用客户自己的工作标准溶液,指标也可根据用户使用要求而定;
3、本技术指标仅供参考,不作为合格性判断依据。
6 校准条件
实验室环境
仪器室内不得有强烈的机械振动和电磁干扰,不得存放与实验无关的易燃、易爆和强腐蚀性气体或试剂;
实验室温度:(15~27) ℃;
相对湿度:≤75%;
标准物质和试剂
八氟萘-异辛烷溶液标准物质,100 pg/μL。不确定度要求
苯甲铜-异辛烷溶液标准物质,10 n g/μL。不确定度要求
六氯苯-异辛烷溶液标准物质,10 n g/μL。不确定度要求
硬脂酸钾脂-异辛烷测试溶液,10 n g/μL。不确定度要求
异辛烷或正己烷,液相色谱级或同等级别。
校准设备
微量注射器,10 μL。
气相色谱仪检定专用测量仪。
7 校准项目和校准方法
仪器不能有影响校准的外观缺陷,按键开关、调节旋钮等各部件工作正常。分辨力
仪器稳定后,执行Autotune命令进行自动调谐,直到调谐通过,打印调谐报告,得到半峰宽W1/2。
注:1. 调谐通常使用的样品为全氟三丁胺 (EC-43)。校准条件标准物质和试剂中没有,纯度有没有要求
2. 也可采用手动谐通。
3. 对于不能打印调谐报告的仪器,可根据调谐结果测量并计算半峰
宽W1/2。
质量范围
以全氟三丁胺调谐样品进行调谐,质量数设定达到600以上,观察质量数600以上(含600)的质谱峰。
信噪比
EI源
仪器调谐通过后,参照附录C条件,注入100 pg/μL的八氟萘-异辛烷溶液1 μL,提取m/z=272离子,再现质谱图,根据公式(1)计算S/N。
S/N=H272/H噪声(1)
式中:
H 272——提取离子(m/z )的峰高
H 噪声——基线噪声。
7.4.2 正CI 源
注入10 n g/μL的苯甲铜-异辛烷溶液溶液1 μL,提取m /z =183离子,再现质谱图,根据公式(1)计算S /N 。
负CI 源
注入100 pg/μL的八氟萘-异辛烷溶液1 μL,提取m /z =272离子,再现质谱图,根据公式(1)计算吗S /N 。
质量准确性
注入10 n g/μL的硬脂酸钾脂-异辛烷溶液1 μL,记录m /z 74、143、199、255和298等硬脂酸钾脂主要离子的实测质量数,有效数值保留到小数点后两位,理论值见附录 E ,根据公式(2)计算实测值与理论值之差,以此评价质量准确性。
ΔM =测i M -
-理i M (2)
式中:
测i M -——第i 个离子三次测量平均值,u ;
理i M ——第i 个离子理论值,u ; 注:1、以最高点及其左右两点的三次扫描所得到的质量数平均值作为实测结果;
2、以实测值与理论值之差绝对值最大的一个作为评价质量准确性数据。 测量重复性
参照 条件,前面例如 注入 μL 浓度为 ng/μL 的六氯苯-异辛烷溶液,连续六次,提取六氯苯特征离子m /z =284,再现质量色谱图,按质量色谱峰进行面积积分,根据公式(4)计算RSD :
16)(RSD 6
12
--=∑=i i
x x ×1x
×100% (3) 式中:
RSD ——相对标准偏差(%);
x i ——六氯苯第i 次测量峰面积;
x——六氯苯6次测量峰面积算术平均值;
i——测量序号。
注:对于CI 源,可采用相应的测试灵敏度的标准物质进行重复性测量。
气相色谱柱箱温度控制
柱箱温度稳定性
把铂电阻温度计的连线连接到数字多用表(或色谱仪检定专用测量仪)上,然后把温度计的探头固定在柱箱中部,设定柱箱温度为70 ℃。加热升温,待温度稳定后,观察10 min,每变化一个数记录一次,求出数字多用表最大值与最小值所对应的温度差值,其差值与10 min 内温度测量的算术平均值的比值,即为柱箱温度稳定性。
程序升温重复性
按的校准条件和方法进行程序升温重复性校准。选定初温50 ℃,终温200 ℃。升温速率10 ℃/min 左右。待初温稳定后,开始程序升温,每分钟记录数据一次,直至终温稳定。此实验重复2~3 次,求出相应点的最大相对偏差(Rd),其值应≤2%,结果按下式计算。
式中:
t max ——相应点的最大温度(℃);
t min——相应点的最小温度(℃);
t——相应点的平均温度(℃)。
谱库检索
根据质量准确性测试总离子流色谱图,得到硬脂酸甲酯质谱图,扣除本底后,在系统提供的谱库内对硬脂酸甲酯进行检索。
8 校准结果处理
根据校准结果,发校准证书,所有校准项目及其结果均应在证书中反映。校准结果的表达按照JJF1071-2000 技术规范的要求,包含标题、实验室名称和地址、送校单位的名称和地址、校准日期、校准所用测量标准的溯源性及有效性说明、校准环境等方面内容。
9 复校时间间隔
台式气相色谱-质谱联用仪的复校时间间隔由用户自定,推荐不超过2 年,更换重要部件、维修或对仪器性能有怀疑时,应随时校准。
附录A 记录格式
附录B 证书格式
附录C 气相色谱和质谱参数
质谱参数
EI 源:
离子化能量:70 eV;
扫描范围:信噪比测试,m/z=200~300;质量准确性测试,m/z=20~350;重复性测试,m/z=200~300;
溶剂延迟:3 min(或视具体情况而定);
离子源和四极杆温度根据厂家推荐值设定;
其它参数,如电子倍增器或光电倍增器工作电压,均以自动或手动调谐时确定的值作为校准参数。
CI 源:
反应气:根据厂家推荐方法选择载气种类和流量;
扫描范围:负化学源信噪比测试,m/z=200~300;正化学源信噪比测试,m/z=100~230;重复性测试,根据测试对象确定;
溶剂延迟:3 min(或视具体情况而定);
离子源和四极杆温度根据厂家推荐值设定;
其它参数,如电子倍增器或光电倍增器工作电压,均以自动或手动调谐时确定的值作为校准参数。
色谱参数(参考条件)
色谱柱:DB-5MS 30 m×mm×μm,或其它类似色谱柱;
进样口温度:250 ℃;
传输线温度:250 ℃;
程序升温:八氟萘和苯甲酮,70 ℃(2 min)→10 ℃/min→ 220 ℃(5min);
六氯苯和硬脂酸甲酯,150 ℃→10 ℃/min →250℃(5 min);
进样方式:不分流进样;进样量:μL;
载气:高纯氦;
流速: mL/min,恒流或恒压(无恒流控制部件)。
注:当色谱柱不同时,柱箱温度可作相应改变。
附录D 不确定度评定
台式 GC-MS 校准,在考察的各项指标中,主要对信噪比进行不确定度评价,不确定度主要来自:
1、n 次测量相对标准偏差,A 类,记为:u 1;
2、所采用标准物质的不确定度,B 类,记为:u 2。
因此,得到合成标准不确定度u c :
u c=2221u u 2
将合成标准不确定度乘以包含因子k (k =2)得到扩展不确定度U :
U =k ×u c
附录E 硬脂酸甲酯主要离子峰理论值
离子(m/z ) 理论值
74
87
129
143
199
255
267
298
附录F 全氟三丁胺主要离子峰值
质量数(m /z ) 质量数(m /z ) 质量数(m /z )
50 131 265
69 132 314
70 145 326
76 150 352
81 164 376
93 169 414
95 176 415
100 181 426
101 214 464
112 219 502
113 220 503
114 226 614
119 264 615
质谱仪的工作原理
质谱仪是一种分析各种同位素并测量其质量及含量百分比的仪器。当一束带电的原子核通过质谱仪中的电场和磁场时,凡其荷质比不相等的,便被分开。S1和S2为两个狭缝,从离子源引出的离子受到施于S1及S2间的电位差,在通过S1的匀强磁场区。进入磁场时的速度由下式决定:
V2=2(q/m)v (1)正离子在这一磁场中运动时其轨道如图中所示半径为r的圆。当离子走过一半圆而抵达照相底片P时会在它上面留下痕迹。由轨道半径r=mv/qB(见洛仑兹力),得
v=qb^r/m (2)合并(1)、(2)式,消去v,即得
q/m=2V/b2r2
因为V、b及r可直接测量得到,所以如果我们能够用其他方法决定离子所带的电荷q,则由上式便可求出离子的质量。
我们可以用质谱仪将电荷相同而质量不同的离子分开。科学家应用这种仪器在1920年左右发现了同种化学元素的原子其质量可以不相同;这些质量不同的
同一种元素的原子被称为同位素。汤姆逊首先利用电磁场测定电子的荷质比的原理,同样可运用到带正电的离子,从荷质比很容易算出该离子的质量。正离子通常带电量等于一个电子(称为单电荷离子)。但有时也带有两个、三个甚至四个电子电量(称为多电荷离子)。目前应用的质谱仪是非常精确的仪器,它不但可以测量出每种同位素之准确质量,并可测定每种同位素在元素中所占的百分比。如将这种仪器略加修改,也可应用到同位素分离。质谱仪的形式很多,但所应用的主要原理及结构却大同小异。图3-32所示是一台现代用质谱仪的主要装置部分。这装置是在真空中,正离子流自离子源引出经过窄隙S进入一曲圆形之电场C1C2,调节C1C2之间的电压,可选择一定能量之正离子,这些正离子随着电场之形状弯曲90°而进入一个半圆形的匀强磁场中,磁场的方向与图面垂直且指向纸内,进入磁场之正离子受磁力作用而沿半圆形轨道进行。因正离子e/m之大小不同,轨道形成大小不等的半圆。分别落在底片上的不同位置也就是说元素将按其质量大小的顺序而排列,故称之为“质谱”。如果我们分别测出每种粒子的电流。就能从这些电流大小的比例中,得出该种在离子源中被电离的物质的各种同位素的成分比例。它也可以把化合物中的不同物质的离子分开和成分分析。
质谱分析法主要是通过对样品的离子的质荷比的分析而实现对样品进行定性和定量的一种方法。因此,质谱仪都必须有电离装置把样品电离为离子,有质量分析装置把不同质荷比的离子分开,经检测器检测之后可以得到样品的质谱图,由于有机样品,无机样品和同位素样品等具有不同形态、性质和不同的分析要求,所以,所用的电离装置、质量分析装置和检测装置有所不同。但是,
不管是哪种类型的质谱仪,其基本组成是相同的。都包括离子源、质量分析器、检测器和真空系统。本节主要介绍有机质谱仪的基本结构和工作原理。
离子源(Ion source)
离子源的作用是将欲分析样品电离,得到带有样品信息的离子。质谱仪的离子源种类很多,现将主要的离子源介绍如下。
电子电离源(Electron Ionization EI)
电子电离源又称EI源,是应用最为广泛的离子源,它主要用于挥发性样品的电离。图是电子电离源的原理图,由GC或直接进样杆进入的样品,以气体形式进入离子源,由灯丝F发出的电子与样品分子发生碰撞使样品分子电离。一般情况下,灯丝F与接收极T之间的电压为70伏,所有的标准质谱图都是在70 eV下做出的。在70 eV电子碰撞作用下,有机物分子可能被打掉一个电子形成分子离子,也可能会发生化学键的断裂形成碎片离子。由分子离子可以确定化合物分子量,由碎片离子可以得到化合物的结构。对于一些不稳定的化合物,在70 eV的电子轰击下很难得到分子离子。为了得到分子量,可以采用1020 eV 的电子能量,不过此时仪器灵敏度将大大降低,需要加大样品的进样量。而且,得到的质谱图不再是标准质谱图。
离子源中进行的电离过程是很复杂的过程,有专门的理论对这些过程进行解释和描述。在电子轰击下,样品分子可能有四种不同途径形成离子:样品分子被打掉一个电子形成分子离子。
分子离子进一步发生化学键断裂形成碎片离子。
分子离子发生结构重排形成重排离子。
通过分子离子反应生成加合离子。
此外,还有同位素离子。这样,一个样品分子可以产生很多带有结构信息的离子,对这些离子进行质量分析和检测,可以得到具有样品信息的质谱图。
电子电离源主要适用于易挥发有机样品的电离,GC-MS联用仪中都有这种离子源。其优点是工作稳定可靠,结构信息丰富,有标准质谱图可以检索。缺点是只适用于易汽化的有机物样品分析,并且,对有些化合物得不到分子离子。
化学电离源(Chemical Ionization, CI )。
有些化合物稳定性差,用EI方式不易得到分子离子,因而也就得不到分子量。为了得到分子量可以采用CI电离方式。CI和EI在结构上没有多大差别。或者说主体部件是共用的。其主要差别是CI源工作过程中要引进一种反应气体。反应气体可以是甲烷、异丁烷、氨等。反应气的量比样品气要大得多。灯丝发出的电子首先将反应气电离,然后反应气离子与样品分子进行离子-分子反应,并使样品气电离。现以甲烷作为反应气,说明化学电离的过程。在电子轰击下,甲烷首先被电离:
甲烷离子与分子进行反应,生成加合离子:
加合离子与样品分子反应:
生成的XH2+和 X+比样品分子XH多一个H或少一个H,可表示为(M1),称为准分子离子。事实上,以甲烷作为反应气,除(M+1)+之外,还可能出现(M+17)+,(M+29)+等离子,同时还出现大量的碎片离子。化学电离源是一种软电离方式,有些用EI方式得不到分子离子的样品,改用CI后可以得到准分子离子,因而可以求得分子量。对于含有很强的吸电子基团的化合物,检测负离子的灵敏度远高于正离子的灵敏度,因此,CI源一般都有正CI和负CI,可以根据样品情况进行选择。由于CI得到的质谱不是标准质谱,所以不能进行库检索。
EI和CI源主要用于气相色谱-质谱联用仪,适用于易汽化的有机物样品分析。
快原子轰击源(Fast Atomic bombardment, FAB)
是另一种常用的离子源,它主要用于极性强、分子量大的样品分析。其工作原理如图所示:
氩气在电离室依靠放电产生氩离子,高能氩离子经电荷交换得到高能氩原子流,氩原子打在样品上产生样品离子。样品置于涂有底物(如甘油)的靶上。靶材为铜,原子氩打在样品上使其电离后进入真空,并在电场作用下进入分析器。电离过程中不必加热气化,因此适合于分析大分子量、难气化、热稳定性差的样品。例如肽类、低聚糖、天然抗生素、有机金属络合物等。FAB源得到的质谱不仅有较强的准分子离子峰,而且有较丰富的结构信息。但是,它与EI源得到的质谱图很不相同。其一是它的分子量信息不是分子离子峰M,而往往是(M+H)+或(M+Na)+等准分子离子峰;其二是碎片峰比EI谱要少。
FAB源主要用于磁式双聚焦质谱仪。
4.电喷雾源(Electron spray Ionization,ESI)
ESI是近年来出现的一种新的电离方式。它主要应用于液相色谱-质谱联用仪。它既作为液相色谱和质谱仪之间的接口装置,同时又是电离装置。它的主要部件是一个多层套管组成的电喷雾喷咀。最内层是液相色谱流出物,外层是喷射气,喷射气常采用大流量的氮气,其作用是使喷出的液体容易分散成微滴。另外,在喷嘴的斜前方还有一个补助气喷咀,补助气的作用是使微滴的溶剂快速蒸发。在微滴蒸发过程中表面电荷密度逐渐增大,当增大到某个临界值时,
离子就可以从表面蒸发出来。离子产生后,借助于喷咀与锥孔之间的电压,穿
过取样孔进入分析器(见图)。
加到喷嘴上的电压可以是正,也可以是负。通过调节极性,可以得到正或负离子的质谱。其中值得一提的是电喷雾喷嘴的角度,如果喷嘴正对取样孔,则取样孔易堵塞。因此,有的电喷雾喷嘴设计成喷射方向与取样孔不在一条线上,而错开一定角度。这样溶剂雾滴不会直接喷到取样孔上,使取样孔比较干净,不易堵塞。产生的离子靠电场的作用引入取样孔,进入分析器。
电喷雾电离源是一种软电离方式,即便是分子量大,稳定性差的化合物,也不会在电离过程中发生分解,它适合于分析极性强的大分子有机化合物,如蛋白质、肽、糖等。电喷雾电离源的最大特点是容易形成多电荷离子。这样,一个分子量为10000 Da的分子若带有10个电荷,则其质荷比只有1000 Da,进入了一般质谱仪可以分析的范围之内。根据这一特点,目前采用电喷雾电离,可以测量分子量在300000 Da以上的蛋白质。图是由电喷雾电离源得到的肌红蛋白的质谱图:
5.大气压化学电离源(Atmospheric pressure chemical Ionization,APCI) 它的结构与电喷雾源大致相同,不同之处在于APCI喷咀的下游放置一个针
状放电电极,通过放电电极的高压放电,使空气中某些中性分子电离,产生H
3
O+,
N 2+,O
2
+和O+等离子,溶剂分子也会被电离,这些离子与分析物分子进行离子-
分子反应,使分析物分子离子化,这些反应过程包括由质子转移和电荷交换产生正离子,质子脱离和电子捕获产生负离子等。图是大气压化学电离源的示意图:
大气压化学电离源主要用来分析中等极性的化合物。有些分析物由于结构和极性方面的原因,用ESI不能产生足够强的离子,可以采用APCI方式增加离子产率,可以认为APCI是ESI的补充。APCI主要产生的是单电荷离子,所以分析的化合物分子量一般小于1000 Da。用这种电离源得到的质谱很少有碎片离子,主要是准分子离子。
以上两种电离源主要用于液相色谱-质谱联用仪。
由上式可知,在一定的B、V条件下,不同m/z的离子其运动半径不同,这样,由离子源产生的离子,经过分析器后可实现质量分离,如果检测器位置不变(即R不变)、连续改变V或B可以使不同m/z的离子顺序进入检测器,实现质量扫描,得到样品的质谱。图是单聚焦分析器原理图,这种单聚焦分析器可以是180°的(如图),也可以是90°或其它角度的,其形状象一把扇子,因此又称为磁扇形分析器。单聚焦分析结构简单,操作方便但其分辨力很低。不能满足有机物分析要求,目前只用于同位素质谱仪和气体质谱仪。单聚集质谱仪分辨力低的主要原因在于它不能克服离子初始能量分散对分辨力造成的影响。在离子源产生的离子当中,质量相同的离子应该聚在一起,但由于离子初始能量不同,经过磁场后其偏转半径也不同,而是以能量大小顺序分开,即磁场也具有能量色散作用。这样就使得相邻两种质量的离子很难分离,从而降低了分辨力。
为了消除离子能量分散对分辨力的影响,通常在扇形磁场前加一扇形电场,扇形电场是一个能量分析器,不起质量分离作用。质量相同而能量不同的离子经过静电电场后会彼此分开。即静电场有能量色散作用。如果设法使静电场的能量色散作用和磁场的能量色散作用大小相等方向相反,就可以消除能量分散
对分辨力的影响。只要是质量相同的离子,经过电场和磁场后可以会聚在一起。另外质量的离子会聚在另一点。改变离子加速电压可以实现质量扫描。这种由电场和磁场共同实现质量分离的分析器,同时具有方向聚焦和能量聚焦作用,叫双聚焦质量分析器(见图). 双聚焦分析器的优点是分辨力高,缺点是扫描速度慢,操作、调整比较困难,而且仪器造价也比较昂贵。
四极杆分析器(Quadrupole analyzer)
离子从离子源进入四极场后,在场的作用下产生振动,如果质量为m,电荷为e的离子从Z方向进入四极场,在电场作用下其运动方程是:
离子运动轨迹可由方程的解描述,数学分析表明,在a, q取某些数值时,运动方程有稳定的解,稳定解的图解形式通常用a, q参数的稳定三角形表示。(图当离子的a, q值处于稳定三角形内部时,这些离子振幅是有限的,因而可以通过四极场达到检测器。在保持V dc/Vrf不变的情况下改变V rf值,对应于一个V rf值,四极场只允许一种质荷比的离子通过,其余离子则振幅不断增大,最后碰到四极杆而被吸收。通过四极杆的离子到达检测器被检测。改变Vrf 值,可以使另外质荷比的离子顺序通过四极场实现质量扫描。设置扫描范围实际上是设置Vrf值的变化范围。当Vrf值由一个值变化到另一个值时,检测器检测到的离子就会从m1变化到m2,也即得到m1到m2的质谱。
V rf的变化可以是连续的,也可以是跳跃式的。所谓跳跃式扫描是只检测某些质量的离子,故称为选择离子监测(select ion monitoring SIM)。当样品量很少,而且样品中特征离子已知时,可以采用选择离子监测。这种扫描方式灵敏度高,而且,通过选择适当的离子使干扰组份不被采集,可以消除组分间
的干扰。SIM适合于定量分析,但因为这种扫描方式得到的质谱不是全谱,因此不能进行质谱库检索和定性分析。
飞行时间质量分析器(Time of flight analyzer)
飞行时间质量分析器的主要部分是一个离子漂移管。图是这种分析器的原理图。离子在加速电压V作用下得到动能,则有:
式中:m:离子的质量
e:离子的电荷量
V:离子加速电压
离子以速度v进入自由空间(漂移区),假定离子在漂移区飞行的时间为T,漂移区长度为L,则:
由式可以看出,离子在漂移管中飞行的时间与离子质量的平方根成正比。也即,对于能量相同的离子,离子的质量越大,达到接收器所用的时间越长,质量越小,所用时间越短,根据这一原理,可以把不同质量的离子分开。适当增加漂移管的长度可以增加分辨力。
飞行时间质量分析器的特点是质量范围宽,扫描速度快,既不需电场也不需磁场。但是,长时间以来一直存在分辨力低这一缺点,造成分辨力低的主要原因在于离子进入漂移管前的时间分散、空间分散和能量分散。这样,即使是质量相同的离子,由于产生时间的先后,产生空间的前后和初始动能的大小不同,达到检测器的时间就不相同,因而降低了分辨力。目前,通过采取激光脉冲电离方式,离子延迟引出技术和离子反射技术,可以在很大程度上克服上述三个原因造成的分辨力下降。现在,飞行时间质谱仪的分辨力可达20000以上。最高可检质量超过300000Da,并且具有很高的灵敏度。目前,这种分析器已广
泛应用于气相色谱-质谱联用仪,液相色谱-质谱联用仪和基质辅助激光解吸飞行时间质谱仪中。下图是基质辅助激光解吸飞行时间质谱仪原理图:离子阱质量分析器
离子阱的结构如图所示。离子阱的主体是一个环电极和上下两端盖电极,环电极和上下两端盖电极都是绕Z轴旋转的双曲面,并满足r20=2Z20( r0 为环形电极的最小半径,Z0为两个端盖电极间的最短距离)。直流电压U和射频电压Vrf加在环电极和端盖电极之间,两端盖电极都处于地电位。
傅立叶变换离子回旋共振分析器(Fourier transform ion cyclotron resonance analyzer, FTICR)
这种分析器是在原来回旋共振分析器的基础上发展起来的。因此,首先叙述一下离子回旋共振的基本原理。假定质荷比m/e的离子进入磁感应强度为B 的磁场中,由于受磁场力的作用,离子作圆周运动,如果没有能量的损失和增加,圆周运动的离心力和磁场力相平衡,即:
式中为离子运动的回旋频率(单位为弧度/秒)。由式可以看出,离子的回旋频率与离子的质荷比成线性关系,当磁场强度固定后,只需精确测得离子的共振频率,就能准确的得到离子的质量。测定离子共振频率的办法是外加一个射频辐射,如果外加射频频率等于离子共振频率,离子就会吸收外加辐射能量而改变圆周运动的轨道,沿着阿基米德螺线加速,离子收集器放在适当的位置就能收到共振离子。改变辐射频率,就可以接收到不同的离子。但普通的回旋共振分析器扫描速度很慢,灵敏度低,分辨力也很差。傅立叶变换离子回旋共振分析器采用的是线性调频脉冲来激发离子,即在很短的时间内进行快速频
率扫描,使很宽范围的质荷比的离子几乎同时受到激发。因而扫描速度和灵敏度比普通回旋共振分析器高得多。图是这种分析器的结构示意图:分析室是一个立方体结构,它是由三对相互垂直的平行板电极组成,置于高真空和由超导磁体产生的强磁场中。第一对电极为捕集极,它与磁场方向垂直,电极上加有适当正电压,其目的是延长离子在室内滞留时间;第二对电极为发射极,用于发射射频脉冲;第三对电极为接收极,用来接收离子产生的信号。样品离子引入分析室后,在强磁场作用下被迫以很小的轨道半径作回旋运动,由于离子都是以随机的非相干方式运动,因此不产生可检出的信号。如果在发射极上施加一个很快的扫频电压,当射频频率和某离子的回旋频率一致时共振条件得到满足。离子吸收射频能量,轨道半径逐渐增大,变成螺旋运动,经过一段时间的相互作用以后,所有离子都做相干运动,产生可被检出的信号。做相干运动的正离子运动至靠近接收极的一个极板时,吸收此极板表面的电子,当其继续运动到另一极板时,又会吸引另一极板表面的电子。这样便会感生出“象电流”(见图),象电流是一种正弦形式的时间域信号,正弦波的频率和离子的固有回旋频率相同,其振幅则与分析室中该质量的离子数目成正比。如果分析室中各种质量的离子都满足共振条件,那么,实际测得的信号是同一时间内作相干轨道运动的各种离子所对应的正弦波信号的叠加。将测得的时间域信号重复累加,放大并经模数转换后输入计算机进行快速傅立叶变换,便可检出各种频率成分,然后利用频率和质量的已知关系,便可得到常见的质谱图。
利用傅立叶变换离子回旋共振原理制成的质谱仪称为傅立叶变换离子回
旋共振质谱仪(Fourier transform ion cyclotron resonance mass spectrometer),简称FT-MS。FT-MS有很多明显的优点:
① 分辨力极高,商品仪器的分辨可超过1×106,而且在高分辨力下不影响灵敏度,而双聚焦分析器为提高分辨力必须降低灵敏度。同时,FT-MS的测量精度非常好,能达到百万分之几,这对于得到离子的元素组成是非常重要的。
② 分析灵敏度高,由于离子是同时激发同时检测,因此比普通回旋共振质谱仪高4个量级,而且在高灵敏度下可以得到高分辨力。
①可以和任何离子源相联,扩宽了仪器功能。
此外还有诸如扫描速度快,性能稳定可靠,质量范围宽等优点。当然,另一方面,FT-MS由于需要很高的超导磁场,因而需要液氦,仪器售价和运行费用都比较贵。
检测器(Detecter)
质谱仪的检测主要使用电子倍增器,也有的使用光电倍增管。图是电子倍增器示意图。由四极杆出来的离子打到高能打拿极产生电子,电子经电子倍增器产生电信号,记录不同离子的信号即得质谱。信号增益与倍增器电压有关,提高倍增器电压可以提高灵敏度,但同时会降低倍增器的寿命,因此,应该在保证仪器灵敏度的情况下采用尽量低的倍增器电压。由倍增器出来的电信号被送入计算机储存,这些信号经计算机处理后可以得到色谱图,质谱图及其它各种信息。
真空系统(Vacuum system)
为了保证离子源中灯丝的正常工作,保证离子在离子源和分析器正常运行,消减不必要的离子碰撞,散射效应,复合反应和离子-分子反应,减小本底与记忆效应,因此,质谱仪的离子源和分析器都必须处在优于10-5 mbar的真空中才能工作。也就是说,质谱仪都必须有真空系统。一般真空系统由机械真空泵和
扩散泵或涡输分子泵组成。机械真空泵能达到的极限真空度为10-3 mbar,不能满足要求,必须依靠高真空泵。扩散泵是常用的高真空泵,其性能稳定可靠,缺点是启动慢,从停机状态到仪器能正常工作所需时间长;涡轮分子泵则相反,仪器启动快,但使用寿命不如扩散泵。但由于涡轮分子泵使用方便,没有油的扩散污染问题,因此,近年来生产的质谱仪大多使用涡轮分子泵。涡轮分子泵直接与离子源或分析器相连,抽出的气体再由机械真空泵排到体系之外。
以上是一般质谱仪的主要组成部分。当然,若要仪器能正常工作,还必须要供电系统,数据处理系统等,因为没有特殊之处,不再叙述。
这样,一个有机化合物样品,由于其形态和分析要求不同,可以选用不同的电离方式使其离子化,再由质量分析器按离子的m/z将离子分开并按一定顺序排列成谱,经检测器检测即得到样品的质谱。图是α-紫罗酮的质谱图。质谱图的横坐标是质荷比m/z ,纵坐标是各离子的相对强度。通常把最强的离子的强度定为100,称为基峰(图中为m/z=121),其他离子的强度以基峰为标准来决定。对于一定的化合物,各离子间的相对强度是一定的,因此,质谱具有化合物的结构特征。
气相色谱仪使用常识~注意事项
气相色谱仪使用常识-注意事项 安装色谱柱 1.安装拆卸色谱柱必须在常温下。 2.填充柱有卡套密封和垫片密封,卡套分三种,金属卡套,塑料卡套,石墨卡套,安装时不易拧的太紧。垫片式密封每次按装色谱柱都要换新的垫片(岛津色谱是垫片密封)。 3.色谱柱两头是否用玻璃棉塞好。防止玻璃棉和填料被载气吹到检测器中。 4.毛细管色谱柱安装插入的长度要根据仪器的说明书而定,不同的色谱汽化室结构不同,所以插进的长度也不同。需要说明的如果你用毛细管色谱柱采用不分流,汽化室采用填充柱接口这时与汽化室连接毛细管柱不能探进太多,略超出卡套即可。 氢气和空气的比例对FID检测器的影响 氢气和空气的比例应1:10,当氢气比例过大时FID检测器的灵敏度急剧下降,在使用色谱时别的条件不变的情况下,灵敏度下降要检查一下氢气和空气流速。氢气和空气有一种气体不足点火时发出“砰”的一声,随后就灭火,一般当你点火电着就灭,再点还着随后又灭是氢气量不足。 使用TCD检测器 1.氢气做载气时尾气一定要排到室外。 2.氮气做载气桥流不能设大,比用氢气时要小的多。 3.没通载气不能给桥流,桥流要在仪器温度稳定后开始做样前在给。 如何判断FID检测器是否点着火 不同的仪器判断方法不同,有基流显示的看基流大小,没有基流显示的用带抛光面的扳手凑近检测器出口,观察其表面有无水汽凝结。 气相色谱常见故障诊断 气相色谱种类很多,性能也各有差别。主要包括两个系统。即气路系统和电路系统。气路系统主要有压力表、净化器、稳压阀、稳流阀、转子流量计、六通进样阀、进样器、色谱柱、检测器等;电子系统包括各用电部件的稳压电源、温控装置、放大线路、自动进样和收集装置、数据处理机和记录仪等电子器件。 要分析和判断色谱仪的故障所在,就必须要熟悉气相色谱的流程和气、电路这两大系统,特别是构成这两个系统部件的结构、功能。色谱仪的故障是多种多样的,而且某一故障产生的原因也是多方面的,必须采用部分检查的方法,即排除法,才可能缩小故障的围。对于气路系统出的故障,不外乎是各种气体(特别是载气)有漏气的现象、气体不好、气体稳压稳流不好等等。 例如:基线若始终向下漂移,即“电平”值逐渐变小至负数,这极有可能是载气泄漏,那么就要查找各个接头部件是否有漏的现象,若不漏而基线仍漂移,则可能是电路系统的故障。色谱气路上的故障,分析工作者可以找出并排除,但要排除电路上的故障则并非易事,就需要分析工作者有一定的电子线路方面的知识,并且要弄清楚主机接线图和各系统的电原理图(尤其是接线图)。在这些图上清楚的画出了控制单元和被控对象间的关系,具体的标明了各接插件引线的编号和去向,按图去检查电路、找寻故障是非常方便的。
气相色谱仪操作规程
GC9560气相色谱仪操作规程 一、仪器的操作 1.开机:打开氮气总阀门,调节减压器至0.4Mpa。打开氮气-空气发生 器的电源,流量显示为0.4Mpa(指向数字4)。 2.打开电脑和色谱工作站,让色谱工作站处于采样状态。 3.检查气相色谱仪上气压表:在气相色谱仪开机前检查气压表显示。 4.打开气相色谱仪的电源。 5.检查气相色谱仪程序(TVOC)的条件。 /min,温度升至250℃,保持2min。 6.按“起始”键,此时工作状态灯亮起,这时所有加热器的温度由初始设置温度上升至设定温度。 7.当检测器的温度上升至大于150℃时,点击“输入键”开始点火。没有一次性点火成功,请间隔再次点火(点火前,工作站处于采样状态,点火后,电压数应为0至5000UV左右。否则用FIDI机械粗调)。 8.点火成功后,检查TENAX管是否安装好和六通阀是否在取样状态。 9.在准备灯的状态下,将加热炉拉近同时计时60s将六通阀拔到分析状态并按“起始键”程序开温开始,工作站开始分析。热解析3min后推出炉子,把六通
阀拔回取样状态。在检测苯时,不需要进行程序升温。 二、标准曲线的制作(TVOC):液体外标法 1. 定标采样:将管子连接在定标装置上,用微量进样器取标样,取0.05μ g、0.1μg、0.5μg、1.0μg 、2.0μg标准溶液分别注入Tenax-TA吸附管,5min 后取下并封存,以完成标准系列制备。(注意:从低浓度到高浓度:每次做之前要将Tenax-TA管吹干净;检查六通阀是否在取样状态)。 2.样品分析: (1)定标完毕后将管柱装好,在准备灯的状态下,将加热炉拉近同时计时60S将六通阀拔到分析状态并按“起始键”程序升温开始,工作站开始分析。热解析3min后推出炉子,把六通阀拔回取样状态。出图完毕炉温,结束分析。 (2)分析结束后,新建文件夹命名为**标线文件夹,点击“键红点”命名保存,保存在上面的标线文件夹里。载入TVOC标准曲线,进行“重分析”,假如保留时间不匹配,把表拖到左下角,分别改动标准曲线表的保留时间,再“重分析”。 3.建立ID表标准线:首先,打开0.05浓度的图谱,清空ID表(点击“清空”和“清空列表”);再点击“加入标准曲线”,再打开0.1浓度的谱图,点击标准曲线表,拉到左下角对时间(乙苯,对接二甲苯,苯乙烯,邻二甲苯),假如ID表中TVOC的保留时间不匹配的话,更改ID表的保留时间(范围±0.15),点击确认,再加载,以此类推,要输入8个含量(浓度),再检查ID表中的定量方法为“面积法”,“双对数”打钩,选择在甲苯项中“参与峰”。再点击ID表中的计算,计算完毕点击导出、命名,最后点击确定保存在标线文件夹中。 4.分析结束后,点击“键红点”命名保存,例tvoc-1。 5.检查ID表,是否是TVOC标准曲线。若不是,则打开ID表,再点击“载入”则找到一个标准曲线的文件名,打开,再确定。 6.打开分析好的样品谱图,输入采样体积,温度,气压,再点击“重分析”。若有不能识别的物质,则点击ID表对照更改时间在重分析得出计算结果。 三、关机 1.先关闭氢空一体机的电源。 2.结束程序升温,再点击“降温”待柱炉温度小于50℃后才能关机。 3.最后关闭氮气总阀,关闭电源。
气相色谱仪原理(图文详解)
气相色谱仪原理(图文详解) 什么是气相色谱 本章介绍气相色谱的功能和用途,以及色谱仪的基本结构。 气相色谱(GC)是一种把混合物分离成单个组分的实验技术。它被用来对样品组分进行鉴定和定量测定: 基子时间的差别进行分离 和物理分离(比如蒸馏和类似的技术)不同,气相色谱(GC)是基于时间差别的分离技术。 将气化的混合物或气体通过含有某种物质的管,基于管中物质对不同化合物的保留性能不同而得到分离。这样,就是基于时间的差别对化合物进行分离。样品经过检测器以后,被记录的就是色谱图(图1),每一个峰代表最初混合样品中不同的组分。 峰出现的时间称为保留时间,可以用来对每个组分进行定性,而峰的大小(峰高或峰面积)则是组分含量大小的度量。 图1典型色谱图
系统 一个气相色谱系统包括 可控而纯净的载气源.它能将样品带入GC系统进样口,它同时还作为液体样品的气化室色谱柱,实现随时间的分离 检测器,当组分通过时,检测器电信号的输出值改变,从而对组分做出响应 某种数据处理装置图2是对此作出的一个总结。 样品 载气源一^ 进样口一^ 色谱柱一^ 检测器一_ 数据处理」 图2色谱系统 气源 载气必须是纯净的。污染物可能与样品或色谱柱反应,产生假峰进入检测器使基线噪音增大等。推荐使用配备有水分、烃类化合物和氧气捕集阱的高纯载气。见图
钢瓶阀 若使用气体发生器而不是气体钢瓶时,应对每一台GC都装配净化器,并且使气源尽可能靠近仪器的背面。 进样口 进样口就是将挥发后的样品引入载气流。最常用的进样装置是注射进样口和进样阀。注射进样口 用于气体和液体样品进样。常用来加热使液体样品蒸发。用气体或液体注射器穿透隔垫将样品注入载气流。其原理(非实际设计尺寸)如图4所示。
气相色谱仪操作步骤(精)
气相色谱仪操作步骤 1 打开氮气、氢气、空气发生器的电源开关(或氮气钢瓶总阀),调整输出压力稳定在0.4Mpa左右(气体发生器一般在出厂时已调整好,不用再调整)。 2. 打开色谱仪气体净化器的氮气开关转到“开”的位置。注意观察色谱仪载气B的柱前压上升并稳定大约5分钟后,打开色谱仪的电源开关。 3. 设置各工作部温度。TVOC分析的条件设置:(a)柱箱:柱箱初始温度50℃、初始时间10min、升温速率5℃/min、终止温度250℃、终止时间10min; (b)进样器和检测器:都是250℃。苯分析时的色谱条件:(a)柱箱:柱箱初始温度100℃、初始时间0min、升温速率0℃/min、终止温度0℃、终止时间0min; (b)进样器和检测器:都是150℃。 4. 点火:待检测器(按“显示、换档、检测器”可查看检测器温度)温度升到100℃以上后,打开净化器上的氢气、空气开关阀到“开”的位置。观察色谱仪上的氢气和空气压力表分别稳定在0.1Mpa和0.15Mpa左右。按住点火开关(每次点火时间不能超过6~8秒钟)点火。同时用明亮的金属片靠近检测器出口,当火点着时在金属片上会看到有明显的水汽。如果在6~8秒时间内氢气没有被点燃,要松开点火开关,再重新点火。在点火操作的过程中,如果发现检测器出口内白色的聚四氟帽中有水凝结,可旋下检测器收集极帽,把水清理掉。在色谱工作站上判断氢火焰是否点燃的方法:观察基线在氢火焰点着后的电压值应高于点火之前。 5. 打开电脑及工作站A,打开一个方法文件:TVOC分析方法或苯分析方法。显示屏左下方应有蓝字显示当前的电压值和时间。接着可以转动色谱仪放大器面板上点火按钮上边的“粗调”旋钮,检查信号是否为通路(转动“粗调”旋钮时,基线应随着变化)。待基线稳定后进样品并同时点击“启动”按钮或按一下色谱仪旁边的快捷按钮,进行色谱数据分析。分析结束时,点击“停止”按钮,数据即自动保存。 8.关机程序:首先关闭氢气和空气气源,使氢火焰检测器灭火。在氢火焰熄灭后再将柱箱的初始温度、检测器温度及进样器温度设置为室温(20-30℃),待温度降至设置温度
气相色谱仪使用方法及实验操作步骤
液相色谱仪、气相色谱仪、原子吸收分光光度计、红外光谱仪、核磁共振、原子发射光谱等分析仪器 气相色谱仪使用方法及实验操作步骤: A、打开氮气、氢气、空气发生器的电源开关(或氮气钢瓶总阀),调整输出压力稳定在0.4Mpa左右(气体发生器一般在出厂时已调整好,不用再调整)。 B、打开色谱仪气体净化器的氮气开关转到“开”的位置。注意观察色谱仪载气B的柱前压上升并稳定大约5分钟后,打开色谱仪的电源开关。 C、设置各工作部温度。TVOC分析的条件设置:(a)柱箱:柱箱初始温度50℃、初始时间10min、升温速率5℃/min、终止温度250℃、终止时间10min; (b)进样器和检测器:都是250℃。脂肪酸分析时的色谱条件:(a)柱箱:柱箱初始温度140℃、初始时间5min、升温速率4℃/min、终止温度240℃、终止时间15min; (b)进样器温度是260℃,检测器温度是280℃。 D、点火:待检测器(按“显示、换档、检测器”可查看检测器温度)温度升到150℃以上后,打开净化器上的氢气、空气开关阀到“开”的位置。观察色谱仪上的氢气和空气压力表分别稳定在0.1Mpa 和0.15Mpa左右。按住点火开关(每次点火时间不能超过6~8秒钟)点火。同时用明亮的金属片靠近检测器出口,当火点着时在金属片上会看到有明显的水汽。如果在6~8秒时间氢气没有被点燃,要松开点火开关,再重新点火。在点火操作的过程中,如果发现检测器出口白色的聚四氟帽中有水凝结,可旋下检测器收集极帽,把水清理掉。在色谱工作站上判断氢火焰是否点燃的方法:观察基线在氢火焰点着后的电压值应高于点火之前。 E、打开电脑及工作站(通道一分析脂肪酸,通道二分析碘),打开一个方法文件:脂肪酸分析方法或碘分析方法。显示屏左下方应有蓝字显示当前的电压值和时间。接着可以转动色谱仪放大器面板上点火按钮上边的“粗调”旋钮,检查信号是否为通路(转动“粗调”旋钮时,基线应随着变化)。待基线稳定后进样品并同时点击“启动”按钮或按一下色谱仪旁边的快捷按钮,进行色谱数据分析。分析结束时,点击“停止”按钮,数据即自动保存。 F、关机程序:首先关闭氢气和空气气源,使氢火焰检测器灭火。在氢火焰熄灭后再将柱箱的初始温度、检测器温度及进样器温度设置为室温(20-30℃),待温度降至设置温度后,关闭色谱仪电源。最后再关闭氮气。 高效液相色谱 我国药典收载高效液相色谱法项目和数量比较表: 鉴于HPLC应用在药品分析中越来越多,因此每一个药品分析人员应该掌握并应用HPLC。 三、色谱法分类 (3) 四、色谱分离原理 (3) II.基本概念和理论 (5) 一、基本概念和术语 (5) 二、塔板理论 (8)
气相色谱检测器 的分类和工作原理及应用范围
气相色谱检测器的分类和工作原理及应用范围 待测组分经色谱柱分离后,通过检测器将各组分的浓度或质量转变成相应的电信号,经放大器放大后,由记录仪或微处理机得到色谱图,根据色谱图对待测组分进行定性和定量分析。 气相色谱监测器根据其测定范围可分为: 通用型检测器:对绝大多数物质够有响应; 选择型检测器:只对某些物质有响应;对其它物质无响应或很小。 根据检测器的输出信号与组分含量间的关系不同,可分为: 浓度型检测器:测量载气中组分浓度的瞬间变化,检测器的响应值与组分在载气中的浓度成正比,与单位时间内组分进入检测器的质量无关。 质量型检测器:测量载气中某组分进入检测器的质量流速变化,即检测器的响应值与单位时间内进人检测器某组分的质量成正比 目前已有几十种检测器,其中最常用的是热导池检测器、电子捕获检测器(浓度型);火焰离子化检测器、火焰光度检测器(质量型)和氮磷检测器等。 一.检测器的性能指标——灵敏度(高)、稳定性(好)、响应(快)、线性范围(宽) (一)灵敏度——应答值 单位物质量通过检测器时产生的信号大小称为检测器对该物质的灵敏度。 响应信号(R)—进样量(Q)作图,可得到通过原点的直线,该直线的斜率就是检测器的灵敏度,以S 表示: (3) 由此可知:灵敏度是响应信号对进入检测器的被测物质质量的变化率。 气相色谱检测器的灵敏度的单位,随检测器的类型和试样的状态不同而异: 对于浓度型检测器: 当试样为液体时,S的单位为mV·ml/mg,即1mL载气中携带1mg的某组分通过检测器时产生的mV数; 当试样为气体时,S的单位为mV·ml/ml,即1ml载气中携带1ml的某组分通过检测器时产生的mV数; 对于质量型检测器:当试样为液体和气体时,S的单位均为:mV·s/g,即每 秒钟有1g的组分被载气携带通过检测器所产生的mV数。 灵敏度不能全面地表明一个检测器的优劣,因为它没有反映检测器的噪音水平。由于信号可以被放大器任意放大,S增大的同时噪声也相应增大,因此,仅用S不能正确评价检测器的性能。 (二)检测限(敏感度)
试题1 气相色谱仪基本操作及考核标准
试题1 气相色谱仪基本操作 1、任务描述 根据作业指导书的指导,能完成气相色谱仪及其辅助设备的基本操作,能描述气相色谱仪各组分部件及其作用,能解释气相色谱仪的分析流程。要求每个抽查的学生在150分钟的时间内独立完成任务。 2、实施条件 (1)场地:天平室,仪器分析检验室。 (2)仪器、试剂: 仪器:带氢焰离子化检测器的气相色谱仪 (3)考核时量 150分钟 (4)考核标准(见附件)
附件水一质检测验任务单 任务单
附件二作业指导书 气相色谱仪基本操作作业指导书 1.气路的安装与检漏训练 钢瓶与减压阀的连接。 减压阀与气体管道的连接。 气体管道与净化器的连接。 净化器与 型气相色谱仪的连接。 检漏操作:用毛笔将皂液涂于各接头处,看是 否有气泡溢出。若有,则表示漏气;若无, 则表示不漏气。 2.气体的打开与设置训练: 逆时针打开载气(N 2)钢瓶总阀,顺时针调节减压阀“ 形杆”至压力表显示输出压力为0.4MPa 气体出口,按逆时针方向打开净化器开关。 调节载气柱前压。 3.载气的流量测定方法 皂膜流量计测定方法 皂膜流量计 认识皂膜流量计: 用皂膜流量计测量稳流阀不同 圈数下载气的准确流量,并绘 制稳流阀圈数~载气流量曲线。 4.气相色谱仪的基本操作 (本过程教师可采用 现场演示 与 结合的 方式来引导学生操作使用 型气相色 谱仪,同时要求教师在学生完成操 练的过程中 同时简单介绍气相色谱仪各组成部分的作用 ) 气相色谱仪开机、关机 打开或关闭 型气相色谱仪的电源开关与加热开关: 注意:要求必须打开载气并使其通入色谱柱后才能打开仪器电源开关与加热开关,同理, 必须关闭仪器电源开关与加热开关之后才能关载气钢瓶与减压阀。 仪器各温度参数的设置训练 设置柱箱温度90℃; 设置检测器温度 130℃; 设置汽化室温度 150℃。 进样操作训练 进样操作步骤: 用丙酮、乙醇等溶剂清洗微量注射器 15次以上。 用待测溶
气相色谱法基本原理及其应用
安徽建筑大学 现代水分析技术论文 专业:xx级市政工程 学生姓名:xxx 学号:xxx 课题:气相色谱法基本原理及其应用指导教师:xxx xx年xx月xx日
气相色谱法基本原理及其应用 xx (安徽建筑工业学院环境与能源工程学院,合肥,230601) 摘要:气相色谱法是分离混合物中各组分的一种有效的手段,其中气相色谱仪是20世纪50年代末在多数科学家的共同努力下诞生的。本文针对气相色谱法的起源与发展历程、工作原理与特点、在环境水污染物分析领域的应用进行了详细的概述,并列举了饮用水中挥发性有机物的气相色谱检测方法,同时提出了该方法新的发展前景。它的发展已在环境监测、水污染控制领中得到了广泛的应用。 关键词:气相色谱法;发展历程;工作原理;水污染物分析 1.气相色谱法的起源与发展历程 (1)气相色谱法的起源 色谱的发现首先认识到这种分离现象和分离方法大有可为的是俄国的植物学家Tswett。Tswett于1903年在波兰华沙大学研究植物叶子的组成时,将叶绿素的石油醚抽提液倒入装有碳酸钙吸附剂的玻璃管上端,然后用石油醚进行淋洗,结果不同色素按吸附顺序在管内形成一条不同颜色的环带,就像光谱一样。1906年,Tswett在德国植物学杂志上发表的一篇论文中首次把这些彩色环带命名为“色谱图”,玻璃管称为“色谱柱”,碳酸钙称为“固定相”,石油醚称为“流动相”。Tswett开创的方法叫做“液-固色谱法”[1-2],这就是色谱法的起源。 1941年,英国科学家Martin和Synge在研究液-液分配色谱时,预言可以使用气体作流动相,即气-夜色谱法。他们在1941年发表的论文中写到“流动相不一定是液体,也可以是蒸气,如以永久性气体带动挥发性混合物,在色谱柱中通过装有浸透不挥发性溶剂的固体时,可以得到很好的分离”[3]。1950年,Martin和James使用硅藻土助滤剂做载体,硅油为固定相,用气体流动相对脂肪酸进行精细分离,这就是气^液分配色谱的起源。后来,他们在1952年的Biochemical Journal上又连续发表了3篇论文[4-6],叙述了用气相色谱分离低碳数脂肪酸、挥发性胺和吡啶类同系物的方法,这标志着气相色谱法正式进入历史舞台。当时在石油化工的分析中,正当传统的分析方法无能为力时,气相色谱法就像及时雨一样,成为化学分析的得力助手。从此,科学家对气相色谱法的研究逐步展开。 (2)气相色谱法的发展 在历史上,气相色谱法的发展总是和气相色谱仪器的发展密不可分。每一种气相色谱新技术的出现,往往都伴随着气相色谱仪器的改进。因此,了解气相色谱法的发展历史可以从气相色谱仪的发展入手。历史上最早的气相色谱仪1947年由捷克色谱学家Jaroslav Janak发明的。该仪器以C为流动相、杜马测氮管为检测器测定分离开的气体体积。在样品和CA 进入测氮管之前,通过KOH溶液吸收掉CA,按时间记录气体体积的增量。这台仪器虽然简陋,但对当时的气相色谱研究起到了巨大的推动作用。Jaroslav Janak发明的气相色谱仪也有一些明显的不足:它只能测室温下为气体的样品, 样品中的CA不能被测定,而且没有实现自动化。20世纪50年代末,它逐渐被更先进的气相色谱仪所取代。W55年,第一台商品化气相色谱仪诞生,标志着气相色谱仪的发展进入了崭新的时代。 现代气相色谱仪主要由5个系统组成,即气路系统、进样系统、分离系统、温度控制系统与检测记录系统。气路系统与温控系统自气相色谱诞生以来很少有突破性的进展。气路系统主要朝自动化方向发展,20世纪90年代出现了采用电子压力传感器和电子流量控制器,通过计算机实现压力和流量自动控制的电子程序压力流量控制系统,这是气路系统的一大进步[7]。温控系统则基本朝着精细、快速、自动化方向发展。相比之下,进样系统、分离系统与检测记录系统是气相色谱仪的核心组成系统,它们的每一次变革和进步都推动着气相色谱的
气相色谱质谱联用仪技术指标(新)
气相色谱/质谱联用仪技术指标 1.2温度:操作环境15?C~35?C 1.3 湿度:操作状态25~50%,非操作状态5~95% 2.性能指标 2.1质谱检测器 2.1.1具有网络通讯功能,可实现远程操作。结构紧凑,无需冷却水及压缩空气冷却。 2.1.2*侧开式面板,无须取下质谱仪机盖即可进行维护。玻璃窗口可显示离子源类 型,灯丝运行情况和离子源连接状态。需提供彩页证明文件。 2.1.3质量数范围:2-1000amu,以0.1amu递增
2.1.4分辨率:单位质量数分辨 2.1.5质量轴稳定性: 优于0.10amu/48小时 2.1.6灵敏度: EI:全扫描灵敏度(电子轰击源EI):1pg八氟萘(OFN),信/噪比≥ 1400:1 (扫描范围: 50-300amu) 2.1.7*仪器检出限IDL:10fg八氟萘。并提供三份以上现场安装验收报告。 2.1.8最大扫描速率:大于19,000amu/秒 2.1.9动态范围:全动态范围为106 2.1.10选择离子模式检测(SIM)最多可有100组,每组最多可选择60个离子 2.1.11质谱工作站可根据全扫描得到的数据,自动选择目标化合物的特征离子并对其进 行分组,最后保存到分析方法当中,无须手动输入。(AutoSIM) 2.1.12具有全扫描/选择离子检测同时采集功能 2.1.13两根长效灯丝的高效电子轰击源,采用完全惰性的材料制成 2.1.14*离子化能量:5~241.5eV 2.1.15离子化电流:0~315uA 2.1.16离子源温度:独立控温,150~350?C可调 2.1.17*分析器:整体石英镀金双曲面四极杆,独立温控, 106?C ~200?C。非预四极杆 加热。需提供彩页等证明文件。 2.1.18质量分析器前有T-K保护透镜。 2.1.19检测器:三维离轴,检测器。长效高能量电子倍增器 2.1.20真空系统:250升/秒以上分子涡轮泵 2.1.21气质接口温度: 独立控温,100~350℃ 2.1.22TID 痕量离子检测技术,在数据采集的过程中优化信号。 2.1.23自动归一化调谐。 2.1.24EI源可以采用氢气做为载气,CI源可以采用氨气替代甲烷气。 2.1.25具备早期维护预报功能(EMF) 2.1.26可提供质量认证功能(OQ/PV) 2.2 气相色谱仪 2.2.1 主机 2.2.1.1 电子流量控制(EPC):所有流量、压力均可以电子控制,以提高重现性,配有13路电子流量控制; 2.2.1.2 压力调节:0.001psi。 2.2.1.3 大气压力传感器补偿高度或环境变化; 2.2.1.4 程序升压/升流:3阶;
安捷伦A气相色谱仪作业指导书
XXXX 环境监测站 安捷伦7820A 气相色谱仪作业指导书 修改记录 1.目的 为了不断提高和保证全站监测工作质量,规范我站的安捷伦7820A 气相色谱仪操作规程,方便分析人员使用、维护仪器。 2.适用范围 此作业指导书适用于安捷伦7820A 气相色谱仪。 3.操作程序
3.1 开机: 3.1.1.打开气源(按相应的检测器所需气体,FID需要氮气、氢气和空气)。 3.1.2打开计算机,进入Windows界面。 3.1.3打开7820A GC电源开关。 3.1.4待仪器自检完毕,双击“联机”图标,进入化学工作站,化学工作站自动与7820A通讯,建立连接。 3.2 7820A配置编辑 3.2.1点击“配置”按钮。在“其他”项目中选择压力单位。 3.1.2柱参数设定点击“色谱柱”按钮,进入柱参数设定画面。点击前面的数字,对该柱的名称、长度、内径、膜厚、最高使用温度、最低使用温度和该柱的类型进行设置;点击该柱下拉式箭头选择连接的进样口,检测器及加热类型;用“↑”和“↓”在各柱之间进行切换。 3.1.3在“模块”项目中选择后进样口和后检测器尾吹气的种类。 4.在“ALS”项目中输入所用自动进样针的规格。 3.3 测试以及数据采集方法编辑: 3.3.1 开始编辑完整方法 从“文件”菜单中选择“新建”→“方法”→“确定”。
3.3.2填写自动进样器的参数: 点击“”,设置进样体积:0.2uL,溶剂A 清洗,进样前清洗4次,进样后清洗4次,体积为最大,溶剂B清洗,进样前清洗4次,进样后清洗4次,体积为最大,样品清洗2次,样品抽吸次数6次,驻留时间,进样前:0分钟,进样后:0分钟,推杆速度:快速,粘度延迟:0秒,采样深度:不启用,进样类型:标准 L1气隙 0.2uL。 注:上述设置是常用设置,对于不同性质的样品,需要对某些参数进行更改,比如对于粘度较大的样品,需要将进样后驻留时间设为3-5s,同时将粘度延迟设为3-5s 3.3.3填写进样口参数: 点击“前进样器”或“后进样器”,根据需要填写前进样口或后进样口参数。输入数值后,在各参数前面打钩。根据需要设置进样口温度、进样的模式(分流、不分流、脉冲分流和脉冲不分流,毛细管柱一般要分流,填充柱一般不分流)。载气节省一般要开启。
气相色谱操作规范流程
安捷伦7890B气相色谱仪-GC-001操作规程 1.仪器分析原理 气相色谱仪是以气体作为流动相(载气),当样品由微量注射器“注射”进入进样器,在衬管中快速挥发后被载气携带进入填充柱或毛细管色谱柱。由于样品中各组份在色谱柱中的流动相(气相)和固定相(液相或固相)间分配或吸附系数的差异,在载气的冲洗下,各组份在两相间作反复多次分配,使各组份在柱中得到分离,然后用接在柱后的检测器根据组份的物理化学特性,将各组份按顺序检测出来。 图1 MDI-100产品异构体色谱图 2.仪器组成及各部件作用 2.1气相色谱仪主要由载气系统、进样系统、分离系统(色谱柱系统)、检测系统、记录系统等五大系统组成。各系统的作用分别为: 载气系统――提供洁净的具有一定流速的载气(流动相)。 进样系统――样品在此气化后进入到色谱柱。 分离系统――分离样品中的各组分。 检测系统――将分离后由色谱柱流出的组分的浓度或质量转变为相应的电信号。 记录系统――将检测到的电信号经处理后记录、并显示。
2.2各部件介绍: 图2 Agilent 7890B GC-001 的前视图 2.2.1进样口 进样口是将样品注射到GC 中的位置。Agilent 7890B GC 最多可以有两个进样口,标为Front Inlet(前进样口)和Back Inlet(后进样口)。GC-001前后进样口为分流/ 不分流进样口 图3 Agilent 7890B GC-001 的前后进样口 2.2.2自动进样器
带有样品盘的Agilent 7683B 自动液体进样器将会自动处理液体样品。Agilent 7890B GC 最多可以有两个自动进样器,标为Front Injector(前进样器)和Back Injector (后进样器)。 2.2.3色谱柱和柱箱 GC 色谱柱位于温度控制柱箱的内部。通常,色谱柱的一端连接进样口,另一端连接检测器。色谱柱因长度、直径和内涂层而异。每个色谱柱被设计为可以处理不同化合物。色谱柱和柱箱的用途是将注入的样品在经过色谱柱时分离成各种化合物。要协助此过程,可以对GC 进行编程,以加速样品流过色谱柱。GC-001前后色谱柱均采用HP-5色谱柱 图4 Agilent 7890B GC-001 柱箱视图 2.2.4检测器 当化合物流出色谱柱时,检测器用于测定其是否存在。当每种化合物进入检测器时,会产生与已检测到的化合物的量成比例的电子信号。此信号通常会被发送到数据分析系统-如EZchrome OpenLab -信号是以色谱图上峰的形式出现在系统中。Agilent 7890B GC-001容纳两个检测器,分别标为Front Det (前检测器)、Back Det (后检测器)。前后检测器均为FID检测器。
气相色谱仪操作规程完全版
气相色谱仪操作规程 GC9790气相色谱仪操作规程(一) (1) SP1000气相色谱仪操作规程 (1) Agilent4890D气相色谱仪操作规程 (2) HP-5890A气相色谱仪操作规程 (3) GC-9790气相色谱仪操作规程(二) (4) SP2100气相色谱仪操作规程 (5) GC-920色谱操作规程 (5) Agilent6890气相色谱仪操作规程 (6) GC9800TT型气相色谱仪操作步骤 (7) GC9800FF型气相色谱仪操作步骤 (8) 9001型气相色谱仪操作规程 (10) SP6800A气相色谱仪的操作说明 (12) GC-930色谱操作规程 (13) GC112A气相色谱操作规程 (14) GC122气相色谱操作规程 (14) GC1690气相色谱仪说明书 (15) 惠普4890D型气相色谱仪标准操作程序 (16) HP6890气相色谱仪操作规程 (19) SP-6890气相色谱仪操作规程 (20) HP-5890A气相色谱仪操作规程 (21) GC-14A气相色谱仪操作规程 (23) HP4890D气相色谱仪操作说明(二) (24) GC9890气相色谱仪操作步骤 (25) 岛津气相色谱GC-2010操作规程 (26) 岛津GC-14CPFID气相色操作规程 (27) GC-14C气相色谱简易操作规程 (27) Agilent6820-GC(ForCerityNDS) (29) 瓦里安CP3800气相色谱操作规程 (33) 安捷伦GC-6820使用规程 (35)
GC9790气相色谱仪操作规程(一) 1.检查仪器电源线连接是否正常、气路管线连接是否正常。 2.打开载气(N2)钢瓶总阀,并调节减压阀开关,使得输出的载气压力在0.3~0.5Mpa之间。 3.调节仪器上的载气调压阀,使得柱前压处在分析工作所需要的压力(一般来说,柱前压在0.05~0.1Mpa之间)。 4.打开电源开关,根据分析要求设置柱温、汽化温度、检测温度等参数,按确定键后仪器升温。同时打开色谱工作站电源。 5.仪器升温到设置温度后,打开空气发生器电源;同时扭开氢气钢瓶阀门,调节氢气减压阀压力在0.3Mpa左右。 6.调节仪器正面右下侧的针形阀,使空气压力在0.05MPa左右,氢气压力在0.15~0.2MPa之间,用点火枪点着FID的火焰,用玻璃片或铁片等冷的物体靠近检测器的盖帽,有水珠凝结表明点火成功(也可以通过观察工作站所显示的基线是否在点火瞬间开始上升来确定是否点火成功)。 7.将仪器右下侧空气、氢气的针形阀压力都缓慢调节到0.1MPa。 8.待基线稳定后开始分析测试工作。 9.分析工作结束后,可以立即关闭氢气钢瓶总阀以及空气发生器电源。 10.调低各路设定温度,使柱温箱、汽化室、检测器温度下降,待柱箱温度低于70℃即可关闭仪器电源。 11.关闭载气钢瓶上的总阀。清理仪器室的进样针、样品等物品,结束GC9790的操作。 SP1000气相色谱仪操作规程 1仪器组成 1.1气源部分,包括氮气钢瓶,氢气源发生器,空气源发生器。 1.2气相主机,包括氢火焰离子化检测器(FID)。 1.3计算机及C-21色谱数据采集单位组成。 2采样操作步骤 2.1选择合适的色谱柱安装于进样器一端,另一端安装于所用的检测器口。 2.2打开载气钢瓶的总阀及减压阀至0.4-0.5Mpa,确定有载气流量后,打开气相主机电源开关。在面板上按“设定”键进入设定参数界面,设定柱温(恒温、程序升温)、设定进样器温度,设定检测器温度。程序升温包括起始温度、起始时间、升温速率、结束温度、结束时间等。仪器在升温状态中,等待指示灯亮,到达所设状态,就绪指示灯亮,即可进样。2.3打开氢气发生器和空气发生器开关,平衡10分钟。按住气相主机上“点火”钮数秒钟即可。按“状态”键切换到状态界面可观察到信号显示及仪器各部件状态。 2.4打开电脑,双击BF-2002色谱工作站图标进入色谱工作站。
气相色谱仪操作步骤
气相色谱仪操作步骤 1、打开氮气、氢气、空气发生器的电源开关(或氮气钢瓶总阀),调整输出压力稳定在0.4Mpa左右(气体发生器一般在出厂时已调整好,不用再调整)。 2、打开色谱仪气体净化器的氮气开关转到“开”的位置。注意观察色谱仪载气B的柱前压上升并稳定大约5分钟后,打开色谱仪的电源开关。 3、设置各工作部温度。TVOC分析的条件设置:(a)柱箱:柱箱初始温度50℃、初始时间10min、升温速率5℃/min、终止温度250℃、终止时间10min; (b)进样器和检测器:都是250℃。脂肪酸分析时的色谱条件:(a)柱箱:柱箱初始温度140℃、初始时间5min、升温速率4℃/min、终止温度240℃、终止时间15min; (b)进样器温度是260℃,检测器温度是280℃。 4、点火:待检测器(按“显示、换档、检测器”可查看检测器温度)温度升到150℃以上后,打开净化器上的氢气、空气开关阀到“开”的位置。观察色谱仪上的氢气和空气压力表分别稳定在0.1Mpa和0.15Mpa左右。按住点火开关(每次点火时间不能超过6~8秒钟)点火。同时用明亮的金属片靠近检测器出口,当火点着时在金属片上会看到有明显的水汽。如果在6~8秒时间内氢气没有被点燃,要松开点火开关,再重新点火。在点火操作的过程中,如果发现检测器出口内白色的聚四氟帽中有水凝结,可旋下检测器收集极帽,把水清理掉。在色谱工作站上判断氢火焰是否点燃的方法:观察基线在氢火焰点着后的电压值应高于点火之前。 5、打开电脑及工作站(通道一分析脂肪酸,通道二分析碘),打开一个方法文件:脂肪酸分析方法或碘分析方法。显示屏左下方应有蓝字显示当前的电压值和时间。接着可以转动色谱仪放大器面板上点火按钮上边的“粗调”旋钮,检查信号是否为通路(转动“粗调”旋钮时,基线应随着变化)。待基线稳定后进样品并同时点击“启动”按钮或按一下色谱仪旁边的快捷按钮,进行色谱数据分析。分析结束时,点击“停止”按钮,数据即自动保存。 8.关机程序:首先关闭氢气和空气气源,使氢火焰检测器灭火。在氢火焰熄灭后再将柱箱的初始温度、检测器温度及进样器温度设置为室温(20-30℃),待温度降至设置温度后,关闭色谱仪电源。最后再关闭氮气。
气相色谱仪的操作步骤
气相色谱仪的操作步骤 气相色谱仪操作步骤: 1、打开氮气、氢气、空气发生器的电源开关(或氮气钢瓶总阀),调整输出压力稳定在0.4Mpa左右(气体发生器一般在出厂时已调整好,不用再调整)。 2、打开色谱仪气体净化器的氮气开关转到“开”的位置。注意观察色谱仪载气B的柱前压上升并稳定大约5分钟后,打开色谱仪的电源开关。 3、设置各工作部温度。TVOC分析的条件设置:(a)柱箱:柱箱初始温度50℃、初始时间10min、升温速率5℃/min、终止温度250℃、终止时间10min; (b)进样器和检测器:都是250℃。脂肪酸分析时的色谱条件:(a)柱箱:柱箱初始温度140℃、初始时间5min、升温速率4℃/min、终止温度240℃、终止时间15min; (b)进样器温度是260℃,检测器温度是280℃。 4、点火:待检测器(按“显示、换档、检测器”可查看检测器温度)温度升到150℃以上后,打开净化器上的氢气、空气开关阀到“开”的位置。观察色谱仪上的氢气和空气压力表分别稳定在0.1Mpa和0.15Mpa左右。按住点火开关(每次点火时间不能超过6~8秒钟)点火。同时用明亮的金属片靠近检测器出口,当火点着时在金属片上会看到有明显的水汽。如果在6~8秒时间内氢气没有被点燃,要松开点火开关,再重新点火。在点火操作的过程中,如果发现检测器出口内白色的聚四氟帽中有水凝结,可旋下检测器收集极帽,把水清理掉。
在色谱工作站上判断氢火焰是否点燃的方法:观察基线在氢火焰点着后的电压值应高于点火之前。 5、打开电脑及工作站(通道一分析脂肪酸,通道二分析碘),打开一个方法文件:**分析方法。显示屏左下方应有蓝字显示当前的电压值和时间。接着可以转动色谱仪放大器面板上点火按钮上边的“粗调”旋钮,检查信号是否为通路(转动“粗调”旋钮时,基线应随着变化)。待基线稳定后进样品并同时点击“启动”按钮或按一下色谱仪旁边的快捷按钮,进行色谱数据分析。(1)外标法或内标法测,首先进标准品或对照品,然后再进待测品;(2)面积归一法测,首先进溶剂进行空白分析,然后再进待测品。如有异常可用溶剂高温吹柱子,直到基线平稳。分析结束时,点击“停止”按钮,数据即自动保存。 6、关机程序:首先关闭氢气和空气气源,使氢火焰检测器灭火。在氢火焰熄灭后再将柱箱的初始温度、检测器温度及进样器温度设置为室温(20-30℃),待温度降至设置温度后,关闭色谱仪电源。最后再关闭氮气。
气相色谱-质谱联用 原理和应用介绍
气相色谱法-质谱联用 气相色谱法–质谱法联用(英语:Gas chromatography–mass spectrometry,简称气质联用,英文缩写GC-MS)是一种结合气相色谱和质谱的特性,在试样中鉴别不同物质的方法。GC-MS的使用包括药物检测(主要用于监督药物的滥用)、火灾调查、环境分析、爆炸调查和未知样品的测定。GC-MS也用于为保障机场安全测定行李和人体中的物质。另外,GC-MS 还可以用于识别物质中以前认为在未被识别前就已经蜕变了的痕量元素。 GC-MS已经被广泛地誉为司法学物质鉴定的金标方法,因为它被用于进行“专一性测试”。所谓“专一性测试”就是能十分肯定地在一个给定的试样中识别出某个物质的实际存在。而非专一性测试则只能指出试样中有哪类物质存在。尽管非专一性测试能够用统计的方法提示该物质具体是那种物质,但存在识别上的正偏差。 目录 1 历史 2 仪器设备 2.1 GC-MS吹扫和捕集 2.2 质谱检测器的类型 3 分析 3.1 MS全程扫描 3.2 选择的离子检测 3.3 离子化类型 3.3.1 电子离子化 3.3.2 化学离子化 3.4 GC-串联MS 4 应用 4.1 环境检测和清洁 4.2 刑事鉴识 4.3 执法方面的应用
4.4 运动反兴奋剂分析 4.5 社会安全 4.6 食品、饮料和香水分析 4.7 天体化学 4.8 医药 5 参考文献 6 参考书目 7 外部链接 历史用质谱仪作为气相色谱的检测器是上个世纪50年代期间由Roland Gohlke和Fred McLafferty首先开发的。当时所使用的敏感的质谱仪体积庞大、容易损坏只能作为固定的实验室装置使用。 价格适中且小型化的电脑的开发为这一仪器使用的简单化提供了帮助,并且,大大地改善了分析样品所花的时间。1964年,美国电子联合公司(Electronic Associates, Inc. 简称EAI)-美国模拟计算机供应商的先驱在开始开发电脑控制的四极杆质谱仪Robert E. Finnigan的指导下[3]开始开发电脑控制的四极杆质谱仪。到了1966年,Finnigan和Mike Uthe的EAI分部合作售出500多台四极杆残留气体分析仪。1967年,Finnigan仪器公司the (Finnigan Instrument Corporation,简称FIC)组建就绪,1968年初就给斯坦福大学和普渡大学发送了第一台GC/MS的最早雏型。FIC最后重新命名为菲尼根公司(Finnigan Corporation)并且继续持世界GC/MS系统研发、生产之牛耳。 1966年,当时最尖端的高速GC-MS (the top-of-the-line high-speed GC-MS units)单元在不到90秒的时间里,完成了火灾助燃物的分析,然而,如果使用第一代GC-MS至少需要16分钟。到2000年使用四极杆技术的电脑化的GC/MS仪器已经化学研究和有机物分析的必不可少的仪器。今天电脑化的GC/MS仪器被广泛地用在水、空气、土壤等的环境检测中;同时也用于农业调控、食品安全、以及医药产品的发现和生产中。 气质联用色谱是由两个主要部分组成:即气相色谱部分和质谱部分。气相色谱使用毛细管柱,其关键参数是柱的尺寸(长度、直径、液膜厚度)以及固定相性质(例如,5%苯基
Agilent 7890B型气相色谱仪操作规程
无 六.工作程序 1.开机 1.1.开机前准备: 1.2.安装相应色谱柱,检查系统完整性。 1.3.流量的调节,开启钢瓶或减压阀,将氮气压力调节至0.5MPa;开启氢气发生器,保证正常供应氢气。
1.4.检漏,用皂液检查柱及各连接是否漏气。 2.检测 2.1.打开电脑,进入windows界面 2.2.Agilent 7890B气相 2.2.1.打开工作站软件双击7890B 联机,连接Agilent 7890B气相色谱仪,进入工作站界面。 2.2.2编辑参数及方法 点击方法---新建方法---点击方法---编辑完整方法---选择进样方式(自动进样),---设置方法参数(进样器、进样口、色谱柱、柱箱、检测器、配置就绪状态等),建立方法名、系统参数、存储 2.2. 3. 从“方法 2.2.4. 在“ 2.2.5. 2.2.6. 点击“选择 2.2.7. 单击“ 。 2.2.8.柱温箱温度参数设定: 点击“柱箱”图标,进入柱温箱参数设定,选中“柱箱温度开启”并输入初始温度(如40℃),最高柱箱温度;在表格中输入升温程序例如:40℃(2min)----10℃/min----90℃(0min)----15℃/min---170℃(2min),点击“应用”钮。 2.2.9.检测器参数设定: 单击“检测器”图标,进行检测器参数设定,选中加热器、空气流量、氢气燃烧流量、尾吹气流量、火焰并输入适当参数,单击“应用”。 2.2.10.信号参数设定:
点击“信号”图标,进入信号参数设定界面。 选择FID数据采集数率(如20HZ)保存,点击“应用”钮。 2.2.11.点击“序列”菜单中选择“新建序列表”选项编辑所需序列并保存,再在序列---序列参数”选项选择数据存储路径并设置新的计数。 2.2.12.待仪器就绪后,基线平稳,从序列菜单中选择“序列表”,运行序列。 2.3. Agilent 7890B气相+ Agilent7697A顶空 2.3.1打开工作站软件双击GC+HS联机Agilent 7890B气相色谱仪、顶空进样器,进入工作站界面中。 2.3.2. 点击方法 柱箱、 2.3.3. 从“方法 一界面。 2.3.4. 在“ 2.3.5. 2.3.6. 2.3.7. 点击“色谱柱”图标,则该图标对应的参数显示出来,从色谱柱库中选择您的柱子并安装,选择合适的柱前压、流速、线速度(三者只输一个即可),点击“应用”。 2.3.8.进样口参数设定: 单击“进样口”图标,进入进样口设定界面,选择进样口的位置(后);模式---选择分流模式,选择合适的分流比并将“加热器”、“压力”、“隔垫吹扫流量”选中,点击“应用”。 2.3.9.柱温箱温度参数设定: 点击“柱箱”图标,进入柱温箱参数设定,选中“柱箱温度开启”并输入初始温度(如40℃),最高柱箱温度;在表格中输入升温程序例如:40℃(2min)----10℃/min----90℃(0min)----15℃