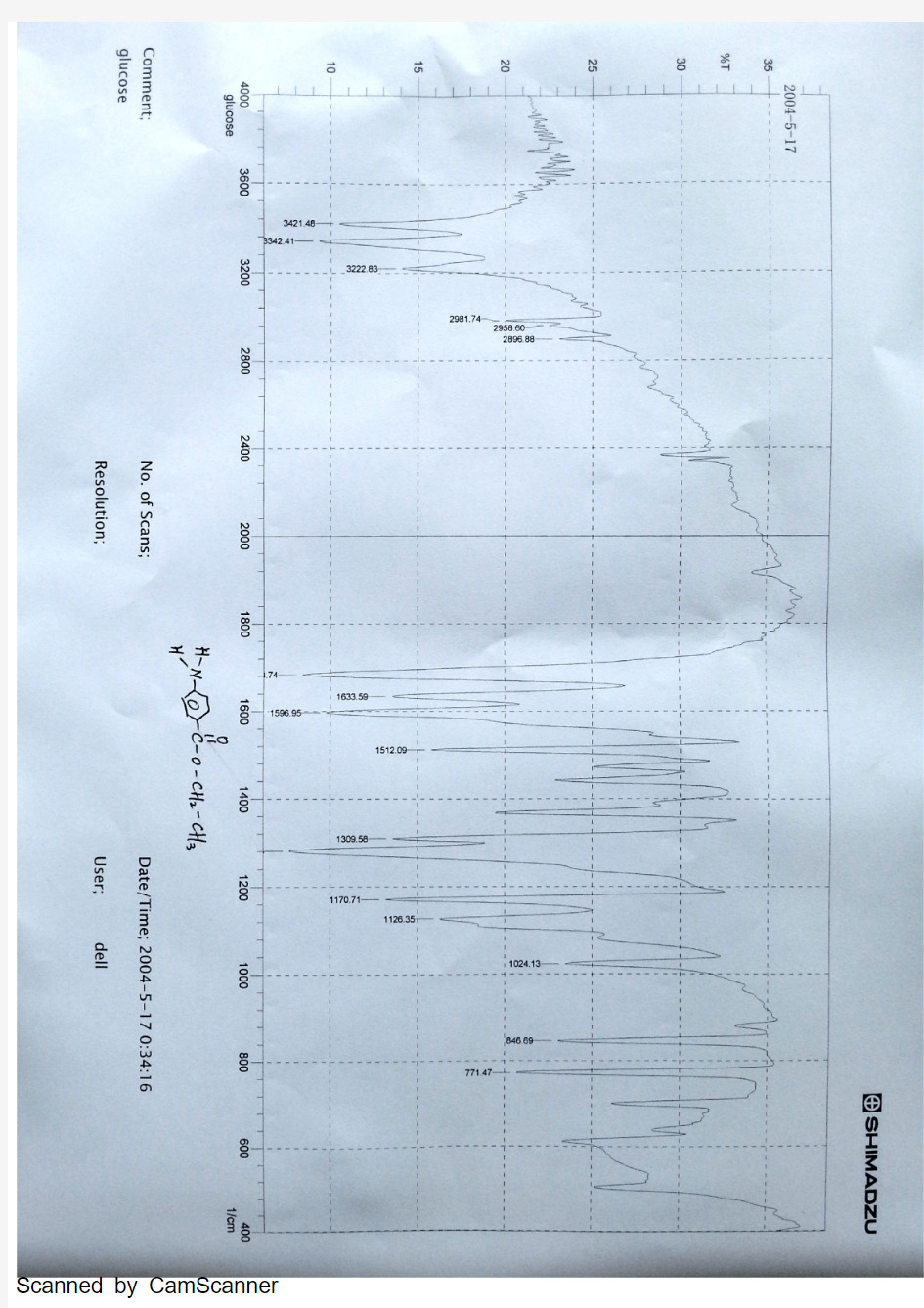
对氨基苯甲酸乙酯红外谱图
Scanned by CamScanner
如何解析红外光谱图解读
如何解析红外光谱图 一、预备知识 (1)根据分子式计算不饱和度公式: 不饱和度Ω=n4+1+(n3-n1)/2其中: :化合价为4价的原子个数(主要是C原子), n 4 :化合价为3价的原子个数(主要是N原子), n 3 n :化合价为1价的原子个数(主要是H,X原子) 1 (2)分析3300~2800cm-1区域C-H伸缩振动吸收;以3000 cm-1为界:高于3000cm-1为不饱和碳C-H伸缩振动吸收,有可能为烯,炔,芳香化合物;而低于3000cm-1一般为饱和C-H伸缩振动吸收; (3)若在稍高于3000cm-1有吸收,则应在 2250~1450cm-1频区,分析不饱和碳碳键的伸缩振动吸收特征峰,其中炔 2200~2100 cm-1,烯 1680~1640 cm-1 芳环 1600,1580,1500,1450 cm-1若已确定为烯或芳香化合物,则应进一步解析指纹区,即1000~650cm-1的频区,以确定取代基个数和位置(顺、反,邻、间、对); (4)碳骨架类型确定后,再依据官能团特征吸收,判定化合物的官能团; (5)解析时应注意把描述各官能团的相关峰联系起来,以准确判定官能团的存在,如2820,2720和1750~1700cm-1的三个峰,说明醛基的存在。 二、熟记健值 1.烷烃:C-H伸缩振动(3000-2850cm-1)C-H弯曲振动(1465-1340cm-1) 一般饱和烃C-H伸缩均在3000cm-1以下,接近3000cm-1的频率吸收。 2.烯烃:烯烃C-H伸缩(3100~3010cm-1),C=C伸缩(1675~1640 cm-1),烯烃C-H 面外弯曲振动(1000~675cm-1)。 3.炔烃:炔烃C-H伸缩振动(3300cm-1附近),三键伸缩振动(2250~2100cm-1)。 4.芳烃:芳环上C-H伸缩振动3100~3000cm-1, C=C 骨架振动1600~1450cm-1, C-H 面外弯曲振动880~680cm-1。 芳烃重要特征:在1600,1580,1500和1450cm-1可能出现强度不等的4个峰。C-H面外弯曲振动吸收880~680cm-1,依苯环上取代基个数和位置不同而发生变化,在芳香化合物红外谱图分析中,常用判别异构体。
origin 处理红外谱图
每年到修改论文的时候,发现很多同学不懂图形和数据处理,出来的图形惨不忍睹。有的人想学没处学,有的根本不想学,最后的结果是研究生给本科生干活,老师给学生干活。所以有空的时候,想以这种方式写一点数据处理技术,给自己的未来减负。先从最简单的单X 多Y图说起。 实验过程中经常遇到系列样品的表征数据,比如红外光谱、X-射线衍射等等,通常这些仪器测定的步长一致,也即X轴完全相同,这时候可以把系列数据绘制在一个图形中,图形的信息量丰富,也方便数据比较。例如这次本科论文中有一位同学的一组红外数据: 先从测试仪器上导出数据,一般都是txt文件,将txt文件直接或经过excel导入到Oringin 软件中,可以通过column/add new column或点击快捷工具来添加多栏数据。
点击作图工具(左下角红色圆圈标示的工具用于线状图),分别设置每一条曲线的X和Y 数据,点击add添加数据。
得到以下的图形: 一般红外光谱图测试范围从4000~400cm-1,为了图形清晰美观,要处理以下几个问题:1)双击X轴数据,出现坐标轴编辑栏,在scale栏下分别编辑X和Y轴的范围和increment(间隔)。 2)点击edit/new layer/top X and right Y,增加一层图形,也就是着增加上X和右Y,这样图形比较方正,这时候还必须在坐标轴编辑栏里将上X和右Y的标尺和数据去掉。在Title&Format里去掉标尺,在Tick Lables里去掉数据。
得到以下的图形: 接下来,要把粘在一起的数据分开,第一步将要移动的数据线激活,对着数据线,点击右键,set as active即可,然后可以采用两种方式移动数据: 1)Anlysis/Subtract/Reference data(减去某个估计的数值)
红外图谱分析方法大全
红外光谱图解析 一、分析红外谱图 (1)首先依据谱图推出化合物碳架类型,根据分子式计算不饱和度。 公式:不饱和度=F+1+(T-O)/2 其中: F:化合价为4价的原子个数(主要是C原子); T:化合价为3价的原子个数(主要是N原子); O:化合价为1价的原子个数(主要是H原子)。 F、T、O分别是英文4,3 1的首字母,这样记起来就不会忘了 举个例子:例如苯(C6H6),不饱和度=6+1+(0-6)/2=4,3个双键加一个环,正好为4个不饱和度。 (2)分析3300~2800cm^-1区域C-H伸缩振动吸收,以3000 cm^-1为界,高于3000cm^-1为不饱和碳C-H伸缩振动吸收,有可能为烯、炔、芳香化合物吗,而低于3000cm^-1一般为饱和C-H伸缩振动吸收。 (3)若在稍高于3000cm^-1有吸收,则应在2250~1450cm^-1频区,分析不饱和碳碳键的伸缩振动吸收特征峰,其中: 炔—2200~2100 cm^-1 烯—1680~1640 cm^-1 芳环—1600、1580、1500、1450 cm^-1 若已确定为烯或芳香化合物,则应进一步解析指纹区,即1000~650cm^-1的频区,以确定取代基个数和位置(顺反,邻、间、对)。 (4)碳骨架类型确定后,再依据其他官能团,如C=O,O-H,C-N 等特征吸收来判定化合物的官能团。 (5)解析时应注意把描述各官能团的相关峰联系起来,以准确判定官能团的存在,如2820、2720和1750~1700cm^-1的三个峰,说明醛基的存在。解析的过程基本就是这样吧,至于制样以及红外谱图软件的使用,一般的有机实验书上都有比较详细的介绍的。 二、记住常见常用的健值 1.烷烃 3000-2850 cm-1C-H伸缩振动 1465-1340 cm-1C-H弯曲振动 一般饱和烃C-H伸缩均在3000 cm-1以下,接近3000 cm-1的频率吸收。 2.烯烃 3100~3010 cm-1烯烃C-H伸缩 1675~1640 cm-1C=C伸缩 烯烃C-H面外弯曲振动(1000~675cm^1)。 3.炔烃 2250~2100 cm-1C≡C伸缩振动 3300 cm-1附近炔烃C-H伸缩振动 4.芳烃 3100~3000 cm-1芳环上C-H伸缩振动 1600~1450 cm-1C=C 骨架振动 880~680 cm-1C-H面外弯曲振动) 芳香化合物重要特征:一般在1600,1580,1500和1450 cm-1可能出现强度不等的4
红外谱图解析基本知识
红外谱图解析基本知识 基团频率区 中红外光谱区可分成4000 cm-1 ~1300(1800)cm-1和1800 (1300 )cm-1 ~ 600 cm-1两个区域。最有分析价值的基团频率在4000 cm-1 ~ 1300 cm-1 之间,这一区域称为基团频率区、官能团区或特征区。区内的峰是由伸缩振动产生的吸收带,比较稀疏,容易辨认,常用于鉴定官能团。 在1800 cm-1 (1300 cm-1 )~600 cm-1 区域内,除单键的伸缩振动外,还有因变形振动产生的谱带。这种振动基团频率和特征吸收峰与整个分子的结构有关。当分子结构稍有不同时,该区的吸收就有细微的差异,并显示出分子特征。这种情况就像人的指纹一样,因此称为指纹区。指纹区对于指认结构类似的化合物很有帮助,而且可以作为化合物存在某种基团的旁证。 基团频率区可分为三个区域 (1) 4000 ~2500 cm-1 X-H伸缩振动区,X可以是O、N、C或S等原子。 O-H基的伸缩振动出现在3650 ~3200 cm-1 范围内,它可以作为判断有无醇类、酚类和有机酸类的重要依据。 当醇和酚溶于非极性溶剂(如CCl4),浓度于0.01mol. dm-3时,在3650 ~3580 cm-1 处出现游离O-H基的伸缩振动吸收,峰形尖锐,且没有其它吸收峰干扰,易于识别。当试样浓度增加时,羟基化合物产生缔合现象,O-H基的伸缩振动吸收峰向低波数方向位移,在3400 ~3200 cm-1 出现一个宽而强的吸收峰。 胺和酰胺的N-H伸缩振动也出现在3500~3100 cm-1 ,因此,可能会对O-H伸缩振动有干扰。 C-H的伸缩振动可分为饱和和不饱和的两种: 饱和的C-H伸缩振动出现在3000 cm-1以下,约3000~2800 cm-1 ,取代基对它们影响很小。如-CH3 基的伸缩吸收出现在2960 cm-1和2876 cm-1附近;R2CH2基的吸收在2930 cm-1 和2850 cm-1附近;R3CH基的吸收基出现在2890 cm-1 附近,但强度很弱。 不饱和的C-H伸缩振动出现在3000 cm-1以上,以此来判别化合物中是否含有不饱和的C-H键。 苯环的C-H键伸缩振动出现在3030 cm-1附近,它的特征是强度比饱和的C-H浆键稍弱,但谱带比较尖锐。 不饱和的双键=C-H的吸收出现在3010~3040 cm-1范围内,末端= CH2的吸收出现在3085 cm-1附近。 叁键oCH上的C-H伸缩振动出现在更高的区域(3300 cm-1 )附近。 (2) 2500~1900 cm-1为叁键和累积双键区,主要包括-CoC、-CoN等叁键的伸缩振动,以及-C =C=C、-C=C=O等累积双键的不对称性伸缩振动。 对于炔烃类化合物,可以分成R-CoCH和R¢-C oC-R两种类型: R-CoCH的伸缩振动出现在2100~2140 cm-1附近; R¢-C oC-R出现在2190~2260 cm-1附近; R-C oC-R分子是对称,则为非红外活性。 -C oN 基的伸缩振动在非共轭的情况下出现2240~2260 cm-1附近。当与不饱和键或芳香核共轭时,该峰位移到2220~2230 cm-1附近。若分子中含有C、H、N原子,-C oN基吸收比较强而尖锐。若分子中含有O原子,且O原子离-C oN基越近,-C oN基的吸收越弱,甚至观察不到。
红外谱图峰位分析方法
红外谱图分析(一) 基团频率和特征吸收峰 物质的红外光谱,是其分子结构的反映,谱图中的吸收峰,与分子中各基团的振动形式相对应。多原子分子的红外光谱与其结构的关系,一般是通过实验手段得到的。这就是通过比较大量已知化合物的红外光谱,从中总结出各种基团的吸收规律来。实验表明,组成分子的各种基团,如O—H、N—H、C—H、C═C、C≡C、C═O等,都有自己特定的红外吸收区域,分子其它部分对其吸收位置影响较小。通常把这种能代表基团存在、并有较高强度的吸收谱带称为基团频率,其所在的位置一般又称为特征吸收峰。 根据化学键的性质,结合波数与力常数、折合质量之间的关系,可将红外4 000~400 cm-1划分为四个区:4 000~2 500 cm-1 氢键区 2 500~2 000 cm-1 产生吸收基团有O—H、C—H、N—H; 叁键区 2 000~1 500 cm-1 C≡C、C≡N、C═C═C 双键区 1 500~1 000 cm-1 C═C、C═O等 单键区 按吸收的特征,又可划分为官能团区和指纹区。 一、官能团区和指纹区 红外光谱的整个围可分成4 000~1 300 cm-1与1 300~600 cm-1两个区域。 4 000~1 300 cm-1区域的峰是由伸缩振动产生的吸收带。由于基团的特征吸收峰一般位于高频围,并且在 该区域,吸收峰比较稀疏,因此,它是基团鉴定工作最有价值的区域,称为官能团区。 在1 300~600 cm-1区域中,除单键的伸缩振动外,还有因变形振动产生的复杂光谱。当分子结构稍有不同时,该区的吸收就有细微的差异。这种情况就像每个人都有不同的指纹一样,因而称为指纹区。指纹区 对于区别结构类似的化合物很有帮助。 指纹区可分为两个波段 (1)1 300~900 cm-1这一区域包括C—O,C—N,C—F,C—P,C—S,P—O,Si—O等键的伸缩振 动和C═S,S═O,P═O等双键的伸缩振动吸收。
常见高分子红外光谱谱图解析
常见高分子红外光谱谱图解析1. 红外光谱的基本原理 1)红外光谱的产生 能量变化 ν νhc h= = E - E = ?E 1 2 ν ν h ?E = 对于线性谐振子 μ κ π ν c 2 1 = 2)偶极矩的变化 3)分子的振动模式 多原子分子振动 伸缩振动对称伸缩 不对称伸缩 变形振动AX2:剪式面外摇摆、面外扭摆、面内摇摆 AX3:对称变形、反对称变形 . 不同类型分子的振动 线型XY2: 对称伸缩不对称伸缩 弯曲
弯曲型XY2: 不对称伸缩对称伸缩面内弯曲(剪式) 面内摇摆面外摇摆卷曲 平面型XY3: 对称伸缩不对称伸缩面内弯曲 面外弯曲 角锥型XY3: 对称弯曲不对称弯曲
面内摇摆 4)聚合物红外光谱的特点 1、组成吸收带 2、构象吸收带 3、立构规整性吸收带 4、构象规整性吸收带 5、结晶吸收带 2 聚合物的红外谱图 1)聚乙烯 各种类型的聚乙烯红外光谱非常相似。在结晶聚乙烯中,720 cm-1的吸收峰常分裂为双峰。要用红外光谱区别不同类型的聚乙烯,需要用较厚的薄膜测绘红外光谱。这些光谱之间的差别反映了聚乙烯结构与线性—CH2—链之间的差别,主要表现在1000-870㎝-1之间的不饱和基团吸收不同,甲基浓度不同以及在800-700㎝-1之间支化吸收带不同。
低压聚乙烯(热压薄膜) 中压聚乙烯(热压薄膜) 高压聚乙烯(热压薄膜)
2.聚丙烯 无规聚丙烯
等规聚丙烯的红外光谱中,在1250-830 cm-1区域出现一系列尖锐的中等强度吸收带(1165、998、895、840 cm-1)。这些吸收与聚合物的化学结构和晶型无关,只与其分子链的螺旋状排列有关。 3.聚异丁烯 CH3 H2 C C n CH3
红外图谱解析
红外图谱解析 首先应该对各官能团的特征吸收熟记于心,因为官能团特征吸收是解析谱图的基础。 对一张已经拿到手的红外谱图: (1)首先依据谱图推出化合物碳架类型:根据分子式计算不饱和度,公式: 不饱和度=F+1+(T-O)/2 其中: F:化合价为4价的原子个数(主要是C原子), T:化合价为3价的原子个数(主要是N原子), O:化合价为1价的原子个数(主要是H原子), F、T、O分别是英文4,3,1的首字母。 举个例子:比如苯:C6H6,不饱和度=6+1+(0-6)/2=4,3个双键加一个环,正好为4个不饱和度; (2)分析3300~2800cm-1区域C-H伸缩振动吸收;以3000 cm-1为界:高于3000cm-1为不饱和碳C-H伸缩振动吸收,有可能为烯, 炔, 芳香化合物,而低于3000cm-1一般为饱和C-H 伸缩振动吸收; (3)若在稍高于3000cm-1有吸收,则应在2250~1450cm-1频区,分析不饱和碳碳键的伸缩振动吸收特征峰,其中: 炔2200~2100 cm-1 烯1680~1640 cm-1 芳环1600,1580,1500,1450 cm-1 若已确定为烯或芳香化合物,则应进一步解析指纹区,即1000~650cm-1的频区,以确定取代基个数和位置(顺反,邻、间、对); (4)碳骨架类型确定后,再依据其他官能团,如C=O, O-H, C-N 等特征吸收来判定化合物的官能团; (5)解析时应注意把描述各官能团的相关峰联系起来,以准确判定官能团的存在,如2820,2720和1750~1700cm-1的三个峰,说明醛基的存在。 解析的过程基本就是这样吧,至于制样以及红外谱图软件的使用,一般的有机实验书上都有比较详细的介绍的,这里就不唠叨了。 这是一个令人头疼的问题,有事没事就记一两个吧: 1.烷烃:C-H伸缩振动(3000-2850cm-1) C-H弯曲振动(1465-1340cm-1)
红外谱图的解析
红外谱图的解析经验 (1)首先依据谱图推出化合物碳架类型:根据分子式计算不饱和度,公式:不饱和度=F+1+(T-O)/2 (2) 分析3300-2800区域C-H伸缩振动吸收;以3000 为界:高于3000为不饱和碳C-H伸缩振动吸收,有可能为烯,炔,芳香化合物,而低于3000一般为饱和C-H伸缩振动吸收; (3)若在稍高于3000有吸收,则应在 2250-1450频区,分析不饱和碳碳键的伸缩振动吸收特征峰,其中:炔 2200-2100,烯 1680-1640,芳环 1600,1580,1500,1450,若已确定为烯或芳香化合物,则应进一步解析指纹区,即1000-650的频区 ,以确定取代基个数和位置(顺反,邻、间、对); (4)碳骨架类型确定后,再依据其他官能团,如 C=O,O-H,C-N 等特征吸收来判定化合物的官能团; (5)解析时应注意把描述各官能团的相关峰联系起来,以准确判定官能团的存在,如2820,2720和1750-1700的三个峰,说明醛基的存在。 1、烷烃:C-H伸缩振动(3000-2850) C-H弯曲振动(1465-1340),一般饱和烃C-H伸缩均在3000以下,接近3000的频率吸收。 2、烯烃:烯烃C-H伸缩(3100-3010) C=C伸缩(1675-1640) 烯烃C-H面外弯曲振动(1000-675)。 3、炔烃:伸缩振动(2250-2100) 炔烃C-H伸缩振动(3300附近)。 4、芳烃:3100-3000, 芳环上C-H伸缩振动 1600-1450, C=C 骨架振动 880-680C-H。 芳香化合物重要特征:一般在1600,1580,1500和1450,可能出现强度不等的4个峰。 880-680,C-H面外弯曲振动吸收,依苯环上取代基个数和位置不同而发生变化 ,在芳香化合物红外谱图分析中,常常用此频区的吸收判别异构体。 5、醇和酚:主要特征吸收是O-H和C-O的伸缩振动吸收, O-H 自由羟基O-H的伸缩振动:3650-3600,为尖锐的吸收峰, 分子间
红外谱图分析方法总结
红外谱图分析方法总结 (1)首先依据谱图推出化合物碳架类型:根据分子式计算不饱和度,公式:不饱和度=F+1+(T-O)/2其中: F:化合价为4价的原子个数(主要是C原子),T:化合价为3价的原子个数(主要是N原子),O:化合价为1价的原子个数(主要是H原子),例如:比如苯:C6H6,不饱和度=6+1+(0-6)/2=4,3个双键加一个环,正好为4个不饱和度;(2)分析3300-2800cm-1区域C-H伸缩振动吸收;以3000cm-1为界:高于3000cm-1为不饱和碳C-H伸缩振动吸收,有可能为烯、炔、芳香化合物,而低于3000cm-1一般为饱和C-H伸缩振动吸收; (3)若在稍高于3000cm-1有吸收,则应在2250-1450cm-1频区,分析不饱和碳碳键的伸缩振动吸收特征峰,其中:炔2200-2100cm-1、烯1680-1640cm-1、芳环1600,1580,1500,1450cm-1。若已确定为烯或芳香化合物,则应进一步解析指纹区,即1000-650cm-1的频区,以确定取代基个数和位置(顺反,邻、间、对);(4)碳骨架类型确定后,再依据其他官能团,如C=O,O-H,C-N等特征吸收来判定化合物的官能团; (5)解析时应注意把描述各官能团的相关峰联系起来,以准确判定官能团的存在,如2820,2720和1750-1700cm-1的三个峰,说明醛基的存在。 至此,分析基本搞定,剩下的就是背一些常见常用的健值了! 1.烷烃:C-H伸缩振动(3000-2850cm-1)C-H弯曲振动(1465-1340cm-1)一般饱和烃C-H伸缩均在3000cm-1以下,接近3000cm-1的频率吸收。 2.烯烃:烯烃C-H伸缩(3100-3010cm-1)C=C伸缩(1675-1640cm-1)烯烃C-H面外弯曲振动(1000-675cm1)。 3.炔烃:伸缩振动(2250-2100cm-1)炔烃C-H伸缩振动(3300cm-1附近)。 4.芳烃:3100-3000cm-1芳环上C-H伸缩振动、1600-1450cm-1C=C骨架振动、880-680cm-1C-H面外弯曲振动、芳香化合物重要特征:一般在1600、1580、1500和1450cm-1可能出现强度不等的4个峰。 880-680cm-1,C-H面外弯曲振动吸收,依苯环上取代基个数和位置不同而发生变化,在芳香化合物红外谱图分析中,常常用此频区的吸收判别异构体。 5.醇和酚:主要特征吸收是O-H和C-O的伸缩振动吸收,O-H自由羟基O-H的伸缩振动:3650-3600cm-1,为尖锐的吸收峰,分子间氢键O-H伸缩振动:3500-3200cm-1,为宽的吸收峰;C-O伸缩振动:1300-1000cm-1O-H面外弯曲:769-659cm-1 6.醚:特征吸收:1300-1000cm-1的伸缩振动,脂肪醚:1150-1060cm-1一个强的吸收峰;芳香醚:两个C-O伸缩振动吸收:1270-1230cm-1(为Ar-O伸缩) 1050-1000cm-1(为R-O伸缩) 7.醛和酮:醛的主要特征吸收:1750-1700cm-1(C=O伸缩)2820,2720cm-1(醛基C-H伸缩);脂肪酮:1715cm-1,强的C=O伸缩振动吸收,如果羰基与烯键或芳环共轭会使吸收频率降低 8.羧酸:羧酸二聚体:3300-2500cm-1宽,强的O-H伸缩吸收1720-1706cm-1,C=O 吸收1320-1210cm-1C-O伸缩,920cm-1成键的O-H键的面外弯曲振动。 9.酯:饱和脂肪族酯(除甲酸酯外)的C=O吸收谱带:1750-1735cm-1区域饱和酯C-C(=O)-O谱带:1210-1163cm-1区域,为强吸收 10.胺:3500-3100cm-1,N-H伸缩振动吸收,1350-1000cm-1,C-N伸缩振动吸收。
红外光谱图解析方法
红外识谱歌 红外可分远中近,中红特征指纹区,1300来分界,注意横轴划分异。 看图要知红外仪,弄清物态液固气。样品来源制样法,物化性能多联系。 识图先学饱和烃,三千以下看峰形。 2960、2870是甲基,2930、2850亚甲峰。 1470碳氢弯,1380甲基显。 二个甲基同一碳,1380分二半。 面内摇摆720,长链亚甲亦可辨。 烯氢伸展过三千,排除倍频和卤烷。 末端烯烃此峰强,只有一氢不明显。 化合物,又键偏,~1650会出现。 烯氢面外易变形,1000以下有强峰。 910端基氢,再有一氢990。 顺式二氢690,反式移至970;单氢出峰820,干扰顺式难确定。 炔氢伸展三千三,峰强很大峰形尖。三键伸展二千二,炔氢摇摆六百八。 芳烃呼吸很特征,1600~1430。1650~2000,取代方式区分明。 900~650,面外弯曲定芳氢。 五氢吸收有两峰,700和750;四氢只有750,二氢相邻830;间二取代出三峰,700、780,880处孤立氢醇酚羟基易缔合,三千三处有强峰。 C-O伸展吸收大,伯仲叔醇位不同。 1050伯醇显,1100乃是仲,1150叔醇在,1230才是酚。 1110醚链伸,注意排除酯酸醇。 若与π键紧相连,二个吸收要看准,1050对称峰,1250反对称。 苯环若有甲氧基,碳氢伸展2820。 次甲基二氧连苯环,930处有强峰,环氧乙烷有三峰,1260环振动,九百上下反对称,八百左右最特征。 缩醛酮,特殊醚,1110非缩酮。 酸酐也有C-O键,开链环酐有区别,开链强宽一千一,环酐移至1250。 羰基伸展一千七,2720定醛基。 吸电效应波数高,共轭则向低频移。 张力促使振动快,环外双键可类比。 二千五到三千三,羧酸氢键峰形宽,920,钝峰显,羧基可定二聚酸、酸酐千八来偶合,双峰60严相隔,链状酸酐高频强,环状酸酐高频弱。 羧酸盐,偶合生,羰基伸缩出双峰,1600反对称,1400对称峰。 1740酯羰基,何酸可看碳氧展。 1180甲酸酯,1190是丙酸,1220乙酸酯,1250芳香酸。 1600兔耳峰,常为邻苯二甲酸。 氮氢伸展三千四,每氢一峰很分明。 羰基伸展酰胺I,1660有强峰;N-H变形酰胺II,1600分伯仲。 伯胺频高易重叠,仲酰固态1550;碳氮伸展酰胺III,1400强峰显。 胺尖常有干扰见,N-H伸展三千三,叔胺无峰仲胺单,伯胺双峰小而尖。 1600碳氢弯,芳香仲胺千五偏。 八百左右面内摇,确定最好变成盐。
origin处理红外谱图
文档来源为:从网络收集整理.word版本可编辑.欢迎下载支持. 每年到修改论文的时候,发现很多同学不懂图形和数据处理,出来的图形惨不忍睹。有的人想学没处学,有的根本不想学,最后的结果是研究生给本科生干活,老师给学生干活。所以有空的时候,想以这种方式写一点数据处理技术,给自己的未来减负。先从最简单的单X 多Y图说起。 实验过程中经常遇到系列样品的表征数据,比如红外光谱、X-射线衍射等等,通常这些仪器测定的步长一致,也即X轴完全相同,这时候可以把系列数据绘制在一个图形中,图形的信息量丰富,也方便数据比较。例如这次本科论文中有一位同学的一组红外数据: 先从测试仪器上导出数据,一般都是txt文件,将txt文件直接或经过excel导入到Oringin 软件中,可以通过column/add new column或点击快捷工具来添加多栏数据。 点击作图工具(左下角红色圆圈标示的工具用于线状图),分别设置每一条曲线的X和Y 数据,点击add添加数据。 得到以下的图形: 一般红外光谱图测试范围从4000~400cm-1,为了图形清晰美观,要处理以下几个问题:1)双击X轴数据,出现坐标轴编辑栏,在scale栏下分别编辑X和Y轴的范围和increment(间隔)。 2)点击edit/new layer/top X and right Y,增加一层图形,也就是着增加上X和右Y,这样图形比较方正,这时候还必须在坐标轴编辑栏里将上X和右Y的标尺和数据去掉。在 Title&Format里去掉标尺,在Tick Lables里去掉数据。 得到以下的图形: 接下来,要把粘在一起的数据分开,第一步将要移动的数据线激活,对着数据线,点击右键,set as active即可,然后可以采用两种方式移动数据: 1)Anlysis/Subtract/Reference data(减去某个估计的数值) 2)Anlysis/Translate/Vertical(垂直移动一个线段),Oringin自动启动“Screen Reader”和“Data Display”两个工具,用鼠标双击图形窗口内的任意两点,曲线就往上或往下移动一段距离。 另外,放入PPT当中的图形曲线不妨用彩色线,但是论文打印稿中一定要用黑色线条。如果论文稿投向国际刊物,所用的英文字体和输出分辨率都有严格的要求,平时处理图像数据时要养成良好的习惯。 Origin是外国人搞的软件,虽然有汉化中文字体,但是在图形中标注中文,往往出现大小不一,或者间隔紊乱的情况,所以还要多采用英文表述。 1文档收集于互联网,如有不妥请联系删除.
Origin 的使用及谱图简单处理
Origin 的使用及谱图简单处理 晁星化学化工学院 061130008 谱图平滑 在红外的测量中,所得到的红外吸收很容易受到一些高频波的影响,如交流电产生的电磁波。这些电磁波会对所得到的红外谱图造成干扰,在图谱解析的时候造成困难,所以需要通过谱图平滑来降低这样的影响,同时又不能破坏图谱所携带的信息。因此在平滑的时候不能仅仅用Origin里的smooth工具直接平滑,这样会造成信息的丢失。 一般使用傅立叶变换(FFT)对谱图进行平滑处理,以去除高频的影响。在特殊的条件下,也可以选取不同频段的信息进行平滑处理。 下图即傅立叶变换前后的醋酸羟基的吸收峰。
图1. FFT平滑前后醋酸羟基的红外吸收峰 分峰处理 在红外光谱、拉曼光谱,甚至是X射线光电子能谱等谱图中都可能需要对重叠的峰进行分峰处理,这样才能确定各个峰的归属,从而判断相应的化学键状态或是化学组成。在分峰时可以使用Gaussian方法和Lorentzian方法。对于交平缓的峰可以使用Gaussian方法进行分峰,如红外中的宽峰。对于较尖锐的峰,则需要用Lorentzian方法进行分峰,如拉曼光谱、X射线光电子能谱。在Origin中可以分别使用这两种方法进行多个峰的拟合,同时也可以自行定义函数,进行两种方法的混合拟合分峰。 以下就是对醋酸羧基部分的分峰处理。由于在醋酸水溶液中,醋酸与水,醋酸与醋酸会形成氢键,从而导致羰基的吸收峰偏移。对羰基部分的吸收峰进行分峰后,就可以帮助判断醋酸水溶液中,醋酸和溶剂相互作用的形式。以下分别采用Gaussian方法和Lorentzian方法处理。可以很容易看出两种方法结果的区别。Gaussian方法需要5个峰才能得到满意的结果,Lorentzian方法可以通过4个峰得到满意的结果。虽然是在红外谱图中,但是由于羰基的峰都是比较尖锐的强吸收,因此使用Lorentzian方法也有其合理性。从而可以得出4种可能较主要的不同的醋酸存在形式。
Origin处理实验数据教学文案
实验 用Origin 软件处理实验数据 实验目的: 了解Origin 软件及其在数据处理中的应用。 实验仪器: 装有Origin 软件的 机一台。 Origin 数据处理软件简介: 数据处理工作是繁琐、枯燥的,值得庆幸的是现在这些工作可以交给计算机来完成。Microcal 软件公司的Origin 软件就是一个短小精悍的数据处理软件。它在Windows 平台下工作,可以完成物理实验常用的数据处理、误差计算、绘图和曲线拟合等工作。这里不对该软件的使用做系统的介绍,只是结合几个例子说明Origin5.0软件在物理实验中经常用到的几项功能。 一、误差计算 前面我们介绍了用千分尺测量钢柱直径的例子,现在用Origin 来处理测量数据。 Origin 中把要完成的一个数据处理任务称做一个“工程”(project )。当我们启动Origin 或在Origin 窗口下新建一个工程时,软件将自动打开一个空的数据表,供输入数据。默认形式的数据表中一共有两列,分别为“A(X)”和“B(Y)”。将下表的8次测 量值输入到数据表的A 列(或B 列)。用鼠标点“A(X)”,选中该列。点“Analysis ”菜单,在下拉菜单项中选“Statistics on Columns ”,瞬间就完成了直径平均值(Mean )、单次测量值的实验标准差)(x S (软件记做sd)、平均值的实验标准差)(x S (软件记做se )的统计计算,其结果如下: 二、绘图
设一小球由静止下落,在不同位置处测量球下落经过的时间,得到数据如下表: 用Origin 软件作图,分析s 与t 之间的关系: 将距离s 的数据输入到A 列,将时间t 的数据输入到B 列,如图二,在“Plot ”下拉菜单中选“Scatter ”,弹出一个对话框。鼠标点“A(X)”,再在右边选“<->X ”,则将“A(X)”设为x 变量。同样,鼠标点“B(Y)”,再在右边选“<->Y”,则将“B(Y)”设为选“Column ”菜单下的“Add New Column ”y 变量。点“OK ” ,出现实验数据的图表,如图三(a)所示。 Origin 默认将图的原点设在第一个数据点的左下方,但是你可以改变这一设置。在“Format ”下拉菜单中点“Axis →X Axis ”,可以修改x 坐标的起止点和坐标示值增量。同样,点“Axis →X Axis ”可以修改y 轴的设置。此外,点“X Axis Titles ” 和“Y Axis Titles ”项可以修改两坐标轴的说明,修改后的一例见图三(b)。 图的右上角有一个文本框,鼠标双击文本框的空白处可以修改框内内容,单击下边工具条上的“T ”按钮,再在图中任意位置点一下,还可以建立一个新的文本框,文本框中可以输入必要的说明。 三、函数图形的绘制 图二 数据表 图三 自由落体的 t -s 图
红外光谱分析
可以按如下步骤来: (1)首先依据谱图推出化合物碳架类型:根据分子式计算不饱和度,公式: 不饱和度=F+1+(T-O)/2 其中: F:化合价为4价的原子个数(主要是C原子), T:化合价为3价的原子个数(主要是N原子), O:化合价为1价的原子个数(主要是H原子), 例如:比如苯:C6H6,不饱和度=6+1+(0-6)/2=4,3个双键 加一个环,正好为4个不饱和度; (2)分析3300~2800cm-1区域C-H伸缩振动吸收;以3000 cm- 1为界:高于3000cm-1为不饱和碳C-H伸缩振动吸收,有可能为烯,炔,芳香化合物,而低于3000cm-1一般为饱和C-H伸缩振动吸收; (3)若在稍高于3000cm-1有吸收,则应在2250~1450cm-1频区,分析不饱和碳碳键的伸缩振动吸收特征峰,其中: 炔 2200~2100 cm-1 烯 1680~1640 cm-1 芳环 1600,1580,1500,1450 cm-1 若已确定为烯或芳香化合物,则应进一步解析指纹区,即 1000~650cm-1的频区,以确定取代基个数和位置(顺反,邻、间、对); (4)碳骨架类型确定后,再依据其他官能团,如 C=O, O-H, C-N 等特征吸收来判定化合物的官能团;
(5)解析时应注意把描述各官能团的相关峰联系起来,以准确判 定官能团的存在,如2820,2720和1750~1700cm-1的三个峰,说明 醛基的存在。 至此,分析基本搞定,剩下的就是背一些常见常用的健值了! 1.烷烃:C-H伸缩振动(3000-2850cm-1) C-H弯曲振动(1465-1340cm-1) 一般饱和烃C-H伸缩均在3000cm-1以下,接近3000cm-1的频率吸收。 2.烯烃:烯烃C-H伸缩(3100~3010cm-1) C=C伸缩(1675~1640 cm-1) 烯烃C-H面外弯曲振动(1000~675cm-1)。 3.炔烃:伸缩振动(2250~2100cm-1) 炔烃C-H伸缩振动(3300cm-1附近)。 4.芳烃:3100~3000cm-1 芳环上C-H伸缩振动 1600~1450cm-1 C=C 骨架振动 880~680cm-1 C-H面外弯曲振动 芳香化合物重要特征:一般在1600,1580,1500和1450cm-1可能出现强度不等的4个峰。 880~680cm-1,C-H面外弯曲振动吸收,依苯环上取代基个数和位置不同而发生变化,在芳香化合物红外谱图分析中,常常用此频区的 吸收判别异构体。 5.醇和酚:主要特征吸收是O-H和C-O的伸缩振动吸收, O-H 自由羟基O-H的伸缩振动:3650~3600cm-1,为尖锐的吸收峰,
Origin-的使用及谱图简单处理
Origin 的使用及谱图简单处理晁星化学化工学院061130008 谱图平滑在红外的测量中,所得到的红外吸收很容易受到一些高频波的影响,如交流电产生的电磁波。这些电磁波会对所得到的红外谱图造成干扰,在图谱解析的时候造成困难,所以需要通过谱图平滑来降低这样的影响,同时又不能破坏图谱所携带的信息。因此在平滑的时候不能仅仅用Origin 里的smooth 工具直接平滑,这样会造成信息的丢失。一般使用傅立叶变换(FFT)对谱图进行平滑处理,以去除高频的影响。在特殊的条件下,也可以选取不同频段的信息进行平滑处理。下图即傅立叶变换前后的醋酸羟基的吸收峰。图1. FFT 平滑前后醋酸羟基的红外吸收峰分峰处理在红外光谱、拉曼光谱,甚至是X 射线光电子能谱等谱图中都可能需要对重叠的峰进行分峰处理,这样才能确定各个峰的归属,从而判断相应的化学键状态或是化学组成。在分峰时可以使用Gaussian 方法和Lorentzian 方法。对于交平缓的峰可以使用Gaussian 方法进行分峰,如红外中的宽峰。对于较尖锐的峰,则需要用Lorentzian 方法进行分峰,如拉曼光谱、X 射线光电子能谱。在Origin 中可以分别使用这两种方法进行多个峰的拟合,同时也可以自行定义函数,进行两种方法的混合拟合分峰。以下就是对醋酸羧基部分的分峰处理。由于在醋酸水溶液中,醋酸与水,醋酸与醋酸会形成氢键,从而导致
羰基的吸收峰偏移。对羰基部分的吸收峰进行分峰后,就可以帮助判断醋酸水溶液中,醋酸和溶剂相互作用的形式。以下分别采用Gaussian 方法和Lorentzian 方法处理。可以很容易看出两种方法结果的区别。Gaussian 方法需要5 个峰才能得到满意的结果,Lorentzian 方法可以通过4 个峰得到满意的结果。虽然是在红外谱图中,但是由于羰基的峰都是比较尖锐的强吸收,因此使用Lorentzian 方法也有其合理性。从而可以得出4 种可能较主要的不同的醋酸存在形式。图2. 醋酸水溶液中羰基吸收峰Gaussian 方法和Lorentzian 方法分峰图谱谱图包装谱图包装也是Origin 的一大功能,包装后的图谱,信息能更好的表现出来,让阅读者一目了然,使图谱更加吸引人。以上的两张图谱也进行了一定的包装。以下的图谱中,对原红外图谱进行了去基线,以及部分放大的处理,所以让图谱更加清晰明了,信息更容易阅读。图3. 谱图包装示例
如何分析红外光谱
如何分析已经拿到手的红外谱图 可以按如下步骤来: (1)首先依据谱图推出化合物碳架类型:根据分子式计算不饱和度,公式:不饱和度=F+1+(T-O)/2 其中: F:化合价为4价的原子个数(主要是C原子), T:化合价为3价的原子个数(主要是N原子), O:化合价为1价的原子个数(主要是H原子), 例如:比如苯:C6H6,不饱和度=6+1+(0-6)/2=4,3个双键加一个环,正好为4个不饱和度; (2)分析3300~2800 cm-1区域C-H伸缩振动吸收;以3000 cm-1 为界:高于3000 cm-1为不饱和碳C-H伸缩振动吸收,有可能为烯,炔,芳香化合物,而低于3000 cm-1一般为饱和C-H伸缩振动吸收; (3)若在稍高于3000 cm-1有吸收,则应在2250~1450 cm-1频区,分析不饱和碳碳键的伸缩振动吸收特征峰,其中: 炔2200~2100 cm-1 烯1680~1640 cm-1 芳环1600,1580,1500,1450 cm-1 若已确定为烯或芳香化合物,则应进一步解析指纹区,即1000~650 cm-1的频区,以确定取代基个数和位置(顺反,邻、间、对); (4)碳骨架类型确定后,再依据其他官能团,如C=O, O-H, C-N 等特征吸收来判定化合物的官能团;
(5)解析时应注意把描述各官能团的相关峰联系起来,以准确判定官能团的存在,如2820,2720和1750~1700 cm-1的三个峰,说明醛基的存在。 至此,分析基本搞定,剩下的就是背一些常见常用的健值了! 1.烷烃:C-H伸缩振动(3000-2850 cm-1) C-H弯曲振动(1465-1340 cm-1) 一般饱和烃C-H伸缩均在3000 cm-1以下,接近3000 cm-1的频率吸收。 2.烯烃:烯烃C-H伸缩(3100~3010 cm-1) C=C伸缩(1675~1640 cm-1) 烯烃C-H面外弯曲振动(1000~675 cm-1)。 3.炔烃:伸缩振动(2250~2100 cm-1) 炔烃C-H伸缩振动(3300 cm-1附近)。 4.芳烃:3100~3000 cm-1 芳环上C-H伸缩振动 1600~1450 cm-1 C=C 骨架振动 880~680 cm-1 C-H面外弯曲振动 芳香化合物重要特征:一般在1600,1580,1500和1450 cm-1可能出现强度不等的4个峰。 880~680 cm-1,C-H面外弯曲振动吸收,依苯环上取代基个数和位置不同而发生变化,在芳香化合物红外谱图分析中,常常用此频区的吸收判别异构体。 5.醇和酚:主要特征吸收是O-H和C-O的伸缩振动吸收,
Origin_使用问题集锦-22个问题
Origin 使用问题集锦 1. 请教怎样反读出 origin 曲线上全部数据点? 如,我用 10个数据点画出了一条 origin 曲线,并存为 project的.OPJ 格式。但,现在我想利用 OPJ 文件从这条曲线上均匀的取出 100个数据点的数值,该如何做?注:要一切都使用 origin 软件完成,不用其他曲线识别软件。 Answer: ORIGIN 中,在分析菜单(或统计菜单)中有插值命令,打开设置对话框,输入数据的起点和终点以及插值点的个数,OK!生成新的插值曲线和对应的数据表格。 2. 如何用origin 做出附件中的图: 其中标注的三角形、方块是怎么整上去的? Answer: 选中左侧竖工具条中的 draw tool(显示是几个点,第七个工具),移动到你要标注的位置双击,就产生了一个点,依次标注完方块。再标注三角的第一个点,标注完后改成三角,以后标注的就都是三角了。改动点的类型的方法和正常画曲线方式一样。 3. 如何用origin 做出附件图中的坐标轴(带刻度)?
Answer: 你把刻度改成那样不就行了。 8.0 的具体方法是双击坐标轴,title & format --> 选左边那个 bottom,然后在右边把 axis 改为 at position=。同理,然后选左边的 left,把axis也改为 at position=。 4. origin能否读取导入曲线的坐标? 一张 bmp 格式的图片,图片内容是坐标系和拟合曲线,但是不知道用什么软件绘制的。请问能否将该图片导入 origin,读出曲线上任意一点的数据? Answer: (1). 1.ORIGIN 有一个图形数字化插件可完成该任务。 2.有许多专门的图形数字化软件也可完成此任务。个人感觉专门的比插件也用、便捷。推荐 WINDIG25 (2). origin下的数字化插件是digitizer,下载地 址:https://www.360docs.net/doc/192227437.html,/fileexchange/details.aspx?fid=8拖入origin即可,但使用不是很方便。比较方便的是un-scan-it。 5. 如何在origin7.5 中标峰值? 用origin7.5 作的XRD图,怎样直接在峰上标数据? Answer: Tools/Pick peaks 设置一下点击 Find Peaks 就 OK了。Positive和Negative 是标正负峰值的意思,其他数值改变一下就知道干吗用的了。 6. 关于origin 拟合曲线延长的问题? 我想把拟合之后的直线向前或向后延长一段距离与坐标轴相交。但是不知道该怎么弄。是不是要改那个范围的最大值和最小值啊?可是怎么改?
如何用Origin处理实验数据
如何用Origin进行数据处理 部分内容摘自以下网址 https://www.360docs.net/doc/192227437.html,/puma0908/blog/item/a3ddfa6d656bfcfe431694ae.html/cmtid/9d3a0e3d5b98e6 ce9f3d62f1 大家可以将实际应用origin时遇到的各种问题及其解决方法写入此文档,对其进行升级。 修改历史 2011-11-25 2011-12-2 1. 数据点的横坐标不是等间距时的曲线绘制(问题提供者:张宇博) 用实验数据作图时,会遇到数据点的横坐标不是等间距的情况,比如: X:1,3,4,8,9,12,... Y:10.2,10.5,11.4,11.8,10.9,10.2,... 如果只有一组实验数据,则按照普通的方法在Worksheet中分别输入X,Y的值,然后用“线+符号”的方式绘图即可。 但是,当有多组此种情况的数据需要绘制在一个图中时,例如: X1:1,3,4,8,9,12,... Y1:10.2,10.5,11.4,11.8,10.9,10.2,... X2:2,5,9,10,11,13,... Y2:13.2,13.5,14.4,13.8,13.9,13.2,... 这时如果将两组数据的X值放在一列里,则Y1和Y2会出现不连续的情况,绘出的曲线发生间断。 解决的办法是: 每组数据的X值都放在各自的X列中,绘出的每条曲线就都是连续的了。具体的操作如图1所示。
第一步:左键双 击此处,弹出下 面的对话框 第二步:将原 来的Y轴指定 为新的X轴 图1 改变数据列的坐标轴属性 2. 多图层下的绘图——图层的使用 1)两组数据的横坐标相差小,纵坐标相差大的情况 2)横坐标相差大,纵坐标相差小的情况 3)横坐标和纵坐标相差都大 图层的建立如图2所示