克莱森Claisen重排
克莱森重排反应
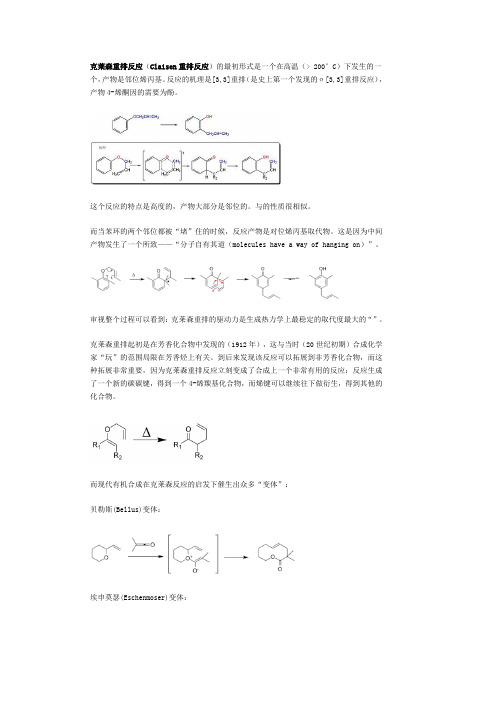
克莱森重排反应(Claisen重排反应)的最初形式是一个在高温(> 200°C)下发生的一个,产物是邻位烯丙基。
反应的机理是[3,3]重排(是史上第一个发现的σ[3,3]重排反应),产物4-烯酮因的需要为酚。
这个反应的特点是高度的,产物大部分是邻位的。
与的性质很相似。
而当苯环的两个邻位都被“堵”住的时候,反应产物是对位烯丙基取代物。
这是因为中间产物发生了一个所致——“分子自有其道(molecules have a way of hanging on)”。
审视整个过程可以看到:克莱森重排的驱动力是生成热力学上最稳定的取代度最大的“”。
克莱森重排起初是在芳香化合物中发现的(1912年),这与当时(20世纪初期)合成化学家“玩”的范围局限在芳香烃上有关。
到后来发现该反应可以拓展到非芳香化合物,而这种拓展非常重要,因为克莱森重排反应立刻变成了合成上一个非常有用的反应:反应生成了一个新的碳碳键,得到一个4-烯羰基化合物,而烯键可以继续往下做衍生,得到其他的化合物。
而现代有机合成在克莱森反应的启发下催生出众多“变体”:贝勒斯(Bellus)变体:埃申莫瑟(Eschenmoser)变体::强生(Johnson)变体:[]天然界的存在在植物代谢的中从到的转换步骤就是一个克莱森重排;该反应受的催化。
预苯酸是一个重要的前体化合物,生物体内含苯环的天然化合物有一大半是由预苯酸转换过来的。
嚬哪醇重排(:pinacol rearrangement)是一个在酸催化下脱水并发生取代基生成羰基化合物的反应。
这一类反应由于(2,3-二甲基-2,3-丁二醇)转换为(3,3-二甲基-2-丁酮)的反应最具代表性,因而得名。
反应的关键步骤是一个的1,2-。
[]反应机理1.两个其中之一接受一个之后脱去一分子水,形成;2.发生1,2-重排,一个基团从未脱去羟基的碳上向有正电荷的碳上转移;3.羟基上脱去一个质子,其氧原子与碳成双键,反应结束。
有机化学重排反应讲解
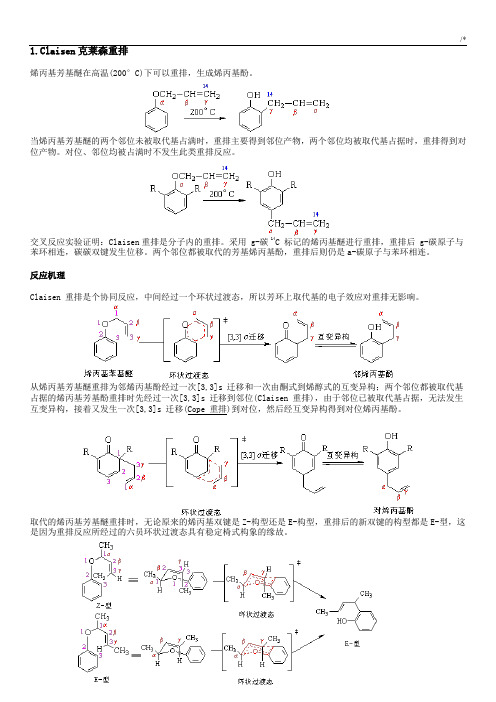
1.Claisen克莱森重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚。
当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要得到邻位产物,两个邻位均被取代基占据时,重排得到对位产物。
对位、邻位均被占满时不发生此类重排反应。
交叉反应实验证明:Claisen重排是分子内的重排。
采用 g-碳 14C 标记的烯丙基醚进行重排,重排后 g-碳原子与苯环相连,碳碳双键发生位移。
两个邻位都被取代的芳基烯丙基酚,重排后则仍是a-碳原子与苯环相连。
反应机理Claisen 重排是个协同反应,中间经过一个环状过渡态,所以芳环上取代基的电子效应对重排无影响。
从烯丙基芳基醚重排为邻烯丙基酚经过一次[3,3]s 迁移和一次由酮式到烯醇式的互变异构;两个邻位都被取代基占据的烯丙基芳基酚重排时先经过一次[3,3]s 迁移到邻位(Claisen 重排),由于邻位已被取代基占据,无法发生互变异构,接着又发生一次[3,3]s 迁移(Cope 重排)到对位,然后经互变异构得到对位烯丙基酚。
取代的烯丙基芳基醚重排时,无论原来的烯丙基双键是Z-构型还是E-构型,重排后的新双键的构型都是E-型,这是因为重排反应所经过的六员环状过渡态具有稳定椅式构象的缘故。
反应实例Claisen 重排具有普遍性,在醚类化合物中,如果存在烯丙氧基与碳碳相连的结构,就有可能发生Claisen 重排。
2.Beckmann贝克曼重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例3.Bamberger,E.重排苯基羟胺(N-羟基苯胺)和稀硫酸一起加热发生重排成对-氨基苯酚:在H2SO4-C2H5OH(或CH3OH)中重排生成对-乙氧基(或甲氧基)苯胺:其他芳基羟胺,它的环上的o-p位上未被取代者会起类似的重排。
醚和环氧化合物化学反应方程式反应路径
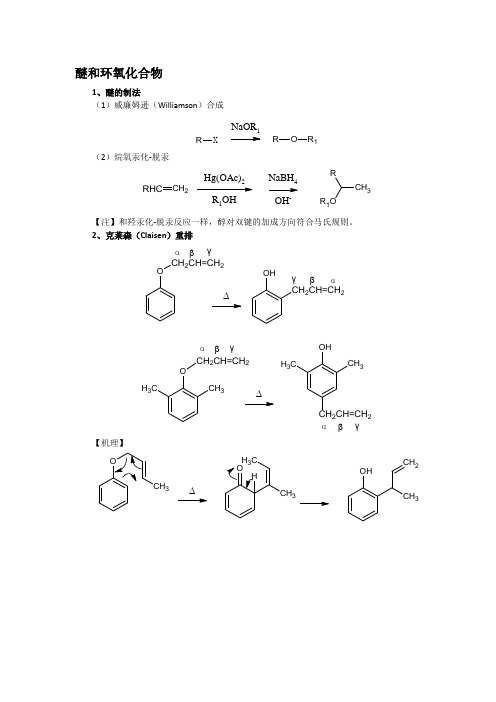
醚和环氧化合物1、醚的制法(1)威廉姆逊(Williamson )合成RXNaOR 1OR 1R(2)烷氧汞化-脱汞RHCCH 2Hg(OAc)2R 1OHNaBH 4OH -CH 3RR 1O【注】和羟汞化-脱汞反应一样,醇对双键的加成方向符合马氏规则。
2、克莱森(Claisen )重排OCH 2CH=CH 2OHCH 2CH=CH 2OCH 2CH=CH 2CH 3CH 3αβγ∆∆OHCH 3CH 3CH 2CH=CH 2αβγαβγαβγ【机理】33∆OHCH 3CH 2CH 3CH 3OCH 2CH=CHC H3α∆OCH 3CHCH =C HC3CH 3OCH 3C3HCHCHCH 2CH2H33αCH 3CH 3OHCH 2CH=CHC H3ααα【注】类似的构型也可发生重排 【例】∆CH 2OOC H 2CH 3CH 3CH 3∆OC3CH 3CH 3OCH 3CH 2∆CH 3O3、冠醚ClOOClOOHOOH+KOH∆OOOOOOOClClOClClOH OHOHOHKOH∆OOOOOO18-冠-6二苯并18-冠-6【特点】冠醚性质最突出就是他有很多醚键,分子中有一定的空穴,金属例子可以钻到空穴中与醚键络合。
OOOOOOK+冠醚分子内圈氧可以与水形成氢键,故有亲水性。
它的外围都是CH2结构,又具有亲油性,因此冠醚能将水相中的试剂包在内圈带到有机相中,从而加速反应,故称冠醚为相转移催化剂。
这种加速非均相有机反应称为相转移催化。
4、环氧化合物(1)开环①酸性开环O CC H+-CCNuOHOCC+【注】不对称环氧化合物的酸性开环方向是亲核试剂优先与取代较多的碳原子结合。
【例】O CH3H +H 3HH 3δ+OH CH 3δ+δ+HOR RO CH 3OH-H +②碱性开环OC2H 5CC OC 2H 5O-HOC 2H 5CC OC 2H 5OH【注】碱性开环,亲核试剂总是先进攻空间位阻较小的,空间效应。
有机化学重排反应 的总结
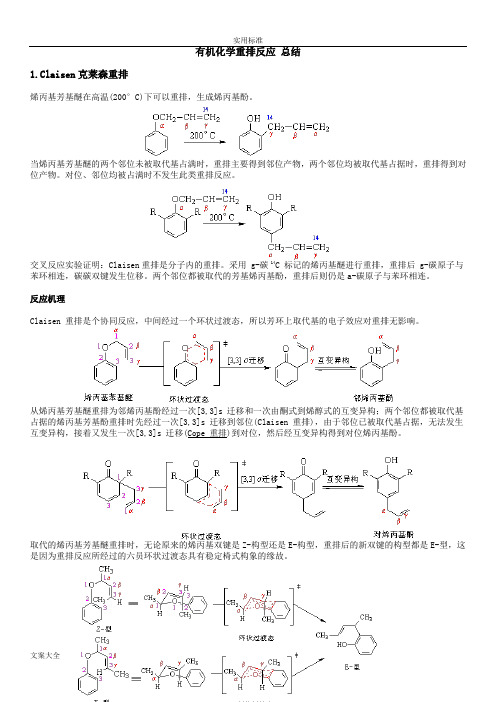
有机化学重排反应总结1.Claisen克莱森重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚。
当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要得到邻位产物,两个邻位均被取代基占据时,重排得到对位产物。
对位、邻位均被占满时不发生此类重排反应。
交叉反应实验证明:Claisen重排是分子内的重排。
采用 g-碳 14C 标记的烯丙基醚进行重排,重排后 g-碳原子与苯环相连,碳碳双键发生位移。
两个邻位都被取代的芳基烯丙基酚,重排后则仍是a-碳原子与苯环相连。
反应机理Claisen 重排是个协同反应,中间经过一个环状过渡态,所以芳环上取代基的电子效应对重排无影响。
从烯丙基芳基醚重排为邻烯丙基酚经过一次[3,3]s 迁移和一次由酮式到烯醇式的互变异构;两个邻位都被取代基占据的烯丙基芳基酚重排时先经过一次[3,3]s 迁移到邻位(Claisen 重排),由于邻位已被取代基占据,无法发生互变异构,接着又发生一次[3,3]s 迁移(Cope 重排)到对位,然后经互变异构得到对位烯丙基酚。
取代的烯丙基芳基醚重排时,无论原来的烯丙基双键是Z-构型还是E-构型,重排后的新双键的构型都是E-型,这是因为重排反应所经过的六员环状过渡态具有稳定椅式构象的缘故。
反应实例Claisen 重排具有普遍性,在醚类化合物中,如果存在烯丙氧基与碳碳相连的结构,就有可能发生Claisen 重排。
2.Beckmann贝克曼重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例3.Bamberger,E.重排苯基羟胺(N-羟基苯胺)和稀硫酸一起加热发生重排成对-氨基苯酚:在H2SO4-C2H5OH(或CH3OH)中重排生成对-乙氧基(或甲氧基)苯胺:其他芳基羟胺,它的环上的o-p位上未被取代者会起类似的重排。
克莱森重排综述

Claisen Rearrangement over the Past Nine DecadesAna M.Martı´n Castro*Departamento de Quı´mica Orga´nica,Universidad Auto´noma de Madrid,Cantoblanco,28049Madrid,SpainReceived February18,2003Contents1.Introduction29392.Definition and Historic Overview of the ClaisenRearrangement29403.Related[3,3]Sigmatropic Rearrangements29413.1.Carroll Rearrangement29413.2.Eschenmoser Rearrangement29413.3.Johnson Rearrangement29423.4.Ireland−Claisen Rearrangement29423.5.Reformatsky−Claisen Rearrangement29423.6.Thio−Claisen Rearrangement29433.7.Aza−Claisen Rearrangement29433.8.Chelate Claisen Rearrangement29433.9.Diosphenol−Claisen Rearrangement29443.10.Metallo−Claisen Rearrangement29443.11.Retro-Claisen Rearrangement29444.Mechanistic and Kinetic Aspects29454.1.General Remarks29454.2.Factors Affecting the Reaction Rate29454.2.1.Influence of the Substituents29454.2.2.Influence of Charged Intermediates29474.2.3.Catalyzed Claisen Rearrangements29494.2.4.Other Parameters29545.Enzymatic Claisen Rearrangement29566.Stereoselective Claisen Rearrangement29566.1.General Aspects29566.2.Intraannular Diastereoselectivity29566.2.1.Transition-State Geometry29566.2.2.Vinyl Double-Bond Geometry29586.2.3.Allyl Double-Bond Geometry29606.2.4.Configuration at C-429616.3.Diastereoselective Synthesis of AchiralProducts29636.3.1.Diastereoselective Synthesis ofCycloalkane Derivatives29636.3.2.E/Z Selectivity29636.4.Diastereoselective Synthesis of ChiralProducts29636.4.1.Chiral Auxiliary at the Allyl Fragment29646.4.2.Chiral Auxiliary at the Vinyl Fragment29656.5.Enantioselective Claisen Rearrangement29686.5.1.Chiral Catalysts29686.5.2.Chiral Solvents29727.Application of Claisen Rearrangement Productsto the Synthesis of Organic Building Blocks29737.1.Heterocyclic Compounds29737.2.Carbocyclic Skeletons29807.3.Dienes29827.4.Condensed Aromatic Structures29847.5.Carboxylic Acid Derivatives29847.6.Quaternary Carbons29887.7.Polysubstituted Alkenes29897.8.Sugar Derivatives29898.Application of Claisen Rearrangement to theSynthesis of Natural Products29909.Other Applications299810.Conclusion299811.Acknowledgments299912.References2999 1.IntroductionThe discovery of the Claisen rearrangement almost a century ago1offered a potentially useful synthetic tool to the organic chemist.Over the decades this usefulness has been realized and the reaction has drawn the attention of numerous research groups, which has been reflected in the number of papers on the topic published in the literature.2In the1970s and1980s several general reviews appeared on the title reaction or related processes.2a-g However,in recent years only specific issues related to this type of rearrangement have been addressed,2k-m the stud-ies on the stereochemical aspects of the reaction deserving special mention.This review provides a general overview covering the most relevant topics related to the Claisen rearrangement,starting from its first publication by Ludwig Claisen in1912as a new[3,3]reorganization of allyl aryl(or vinyl)ethers up to its most recent applications in different organic chemistry fields.First,a brief description of the reaction along with its historic profile are given.This leads to the presentation of other[3,3]rearrange-ments closely related to the title reaction,which are of relevant interest for having been largely exploited as synthetic methods.Next,mechanistic and kinetic aspects are discussed with attention focused on the main factors affecting the reaction rate,basically the presence of different substituents at the1,5-hetero-diene skeleton,the use of catalysts,and changes in the physical parameters affecting the reaction.The following section in the review briefly deals with the enzymatic version of the Claisen rearrangement, which is of relevant interest in metabolic routes.A significant section of the review is constituted by the study of the stereoselective version of the rearrangement.After a presentation of some general*E-mail:martin.castro@uam.es.2939 Chem.Rev.2004,104,2939−300210.1021/cr020703u CCC:$48.50©2004American Chemical SocietyPublished on Web04/23/2004aspects regarding chirality transfer in these pro-cesses,different strategies to control intraannular diastereoselectivity as well as methods to perform diastereoselective synthesis of both achiral and chiral compounds are examined.The enantioselective Clais-en rearrangement is also thoroughly considered.Finally,in the last sections of the review a selection of the most outstanding applications of the reaction is presented.A number of examples illustrating the use of the Claisen rearrangement in the preparation of a wide range of synthetically interesting building blocks and natural or biologically active compounds are considered.Some other potentially interesting applications of the rearrangement in further fields of organic chemistry are also presented.2.Definition and Historic Overview of the Claisen RearrangementThe [3,3]sigmatropic rearrangement of allyl vinyl ethers,which allows the preparation of γ,δ-unsatur-ated carbonyl compounds,is worthy of study due to its special synthetic relevance as well as the large number of theoretical studies generated.This reac-tion,first reported by Ludwig Claisen in 1912,1was originally described as “the thermal isomerization of an allyl vinyl ether 1s or of its nitrogen or sulfur containing analogue derivatives s to afford a bifunc-tionalized molecule 2”(Scheme 1)in a [π2s +σ2s +π2s]process.This first paper essentially described the transfor-mation of allyl phenyl ether into C -allyl phenol.However,it also dealt with the formation,startingfrom O -allylated ethyl acetoacetate (3),of its C -allyl isomer 4after distillation in the presence of NH 4Cl (Scheme 2)in a process which adopted the generaldenomination of Claisen rearrangement .As we shall see,this is a reaction exhibiting all the essential properties required by a synthetic procedure to be considered as efficient:it can be chemo-,regio-,diastereo-,and enantioselective,3can be performed under mild conditions,and affords potentially useful polyfunctionalized molecules.The synthetic potential of the process encouraged,in the following years after its first publication,a number of research groups to make significant efforts to find the experimental conditions which would allow the generalization of the reaction to a wide variety of substrates.To verify that the conditions initially reported by Claisen to perform the rear-rangement on aromatic substrates 4could be success-fully applied to aliphatic skeletons,independently Bergman and Corte in 19355a and Lauer and Kilburn in 19375b studied the rearrangement in the presence of NH 4Cl of ethyl cinnamyl oxycrotonate (5).This substrate was generated either by reaction of cin-namyl alcohol and ethyl 3-ethoxy-2-crotonate 5a or from sodium cinnamylate and ethyl -chlorocrotonate 5b (Scheme 3).The formation of the rearranged productafforded a formal way of S N 2′C -alkylation of cin-namyl halides with the anion derived from aceto-acetates.The interest of this new rearrangement prompted the development of different methods for the prepa-ration of the starting materials.Hurd and Pollack 6described the synthesis of allyl vinyl ethers by acidic or basic elimination as well as the rearrangement of such compounds into the corresponding γ,δ-unsatur-ated carbonyl compounds (Scheme 4).However,this method did not provide general access to allyl vinylethers.Ana M.Martin Castro was born in Madrid,Spain,in 1964.She received her B.S.(1988)and Ph.D.(1994)degrees under the supervision of Professors J.L.Garcia Ruano and J.H.Rodriguez Ramos at the Universidad Auto ´noma of Madrid.As a postdoctoral fellow she joined the groups of Professor P.R.Raithby at the University of Cambridge (U.K.,1997)and Professor V.K.Aggarwal at the University of Sheffield (U.K.,1998−2000).She retuned to Professor Garcia Ruano’s group as an Assistant Professor.Her present research interests include the develop-ment of novel asymmetric methodologies assisted by a chiral sulfinyl group,particularly hydrocyanation processes and Diels −Alder and 1,3-dipolar cycloadditions.Scheme1Scheme2Scheme3Scheme42940Chemical Reviews,2004,Vol.104,No.6Martı´n CastroSeveral years later this procedure for the synthesis of allyl vinyl ethers was improved by the interchange of alcohols with alkyl vinyl ethers catalyzed by Hg(OAc)2.7Those compounds once again proved to be excellent substrates to undergo a [3,3]rearrange-ment (Scheme 5).This mercury-catalyzed reactionhas become one of the typical methods of preparation of allyl vinyl ethers despite that the yields of these reactions are often low.The development of the aliphatic Claisen rear-rangement was simultaneous with the study of the aromatic version of the reaction.2a,b,8Thus,in the Claisen rearrangement of an allyl aryl ether,the first [3,3]step affords an ortho dienone which usually enolizes into an o -allylphenol.It is the reaction known as the ortho Claisen rearrangement (Scheme 6).When the rearrangement takes place on an orthoposition bearing a substituent,a second [3,3]rear-rangement (Cope rearrangement)takes place fol-lowed by enolization.This reaction,usually called the para Claisen rearrangement ,leads to the correspond-ing p -allylphenol.The product resulting from the ortho Claisen rear-rangement is usually obtained from the reaction,although the para process can compete even when both ortho positions are unoccupied.2b The proposed mechanism for the aromatic Claisen rearrangement has been corroborated by the product resulting from the rearrangements of allyl phenyl ether and allyl 2,6-dimethylphenyl ether,both compounds 14C-labeled on the γcarbon of the allyl chain 8a (see Scheme 6).The result of the ortho rearrangement shows that the rearrangement with inversion at the allyl group is the only reaction taking place.In the case of the para rearrangement,the migration only proceeds without inversion of the allyl group.This means that during the course of the reaction such a group is never free enough to undergo resonance.8b3.Related [3,3]Sigmatropic RearrangementsThe interest generated by the Claisen rearrange-ment prompted the development of a considerable number of different versions of [3,3]sigmatropicrearrangement.Some of the most noteworthy ones will be considered next.3.1.Carroll RearrangementThe Carroll reaction,initially described in 1940,9is a thermal rearrangement of allylic -ketoesters followed by decarboxylation 10to yield γ,δ-unsaturated ketones (Scheme 7).This reaction has not beenwidely developed due to the drastic conditions (tem-peratures of 130-220°C after in situ preparation of the -ketoester)which are required to perform the transformation.After the publication of these results,it was re-ported that dianions derived from allylic acetoace-tates,prepared by treatment of acetoacetates with 2equiv of LDA,rearranged under milder thermal con-ditions to give easily isolated -keto acids (Scheme 8).11A dependence of the reaction rate on the substitu-tion pattern on the allylic fragment (R,R ′)H,alkyl,aryl)has also been detected.Thus,acetoacetates derived from primary alcohols rearrange more slowly than those derived from secondary and tertiary alcohols.3.2.Eschenmoser RearrangementIn 1964Eschenmoser,12based on observations previously reported by Meerwein 13on the interchange of amide acetals with allylic alcohols,described the [3,3]rearrangement of N,O -ketene acetals to yield γ,δ-unsaturated amides (Scheme 9).Scheme5Scheme6Scheme7Scheme8Scheme9Claisen Rearrangement over the Past Nine Decades Chemical Reviews,2004,Vol.104,No.62941This reaction allows the formation of a carbon -carbon bond at the position to a nitrogen atom,which is of great applicability in alkaloid synthesis,although it has the inconvenience of the difficulty inherent to the preparation of more elaborated N,O -ketene acetals,which usually requires elevated tem-peratures leading,in some cases,to decomposition of the resulting amides.Several years later an Eschenmoser rearrangement by reaction of lithium allyl alkoxides with acyclic 14and cyclic 15salts of N,N -dialkylalkoxymethylene-iminium was reported to proceed in excellent yields (Scheme 10).The high temperatures reported for theEschenmoser rearrangement are usually required for the alcohol exchange reaction,not for the actual rearrangement.Therefore,the mild conditions em-ployed for the preparation of N,O -ketene acetals such as those depicted in Scheme 10increased the syn-thetic interest of the method.Similar results are obtained by the ynamine -Claisen rearrangement,16also known as Ficini -Claisen rearrangement,by reaction of an allylic alco-hol with 1-(diethylamino)propyne (Scheme 11).Thisis a process whose stereochemical course may be modified by the reaction conditions,as discussed in section 6.2.2.When the N,O -ketene acetal is obtained by adding the alcohol slowly to a refluxing solution of the ynamine in xylene,the rearrangement takes place via the kinetically formed (E )-isomer.In the presence of a Lewis acid,equilibration to the ther-modynamically favored (Z )-stereoisomer occurs before rearrangement.Transformation of the kinetically favored (E )-N,O -ketene acetal to the threo γ,δ-un-saturated amide can be considered as complementary to the Eschenmoser rearrangement,which evolves through the (Z )-isomer affording the erythro product.3.3.Johnson RearrangementFirst reported in 1970,17the Johnson rearrange-ment,which may afford trans -trisubstituted alkenes,was originally described as the process consisting of the heating of an allylic alcohol (6)with an excess of ethyl orthoacetate in the presence of trace amounts of a weak acid (typically propionic acid).The initially formed mixed ortho ester (7)loses ethanol to generate the ketene acetal 8,which undergoes rearrangement leading to a γ,δ-unsaturated ester (9)(Scheme 12).Subsequently the Claisen rearrangement of ortho esters was shown to be compatible with the presence of a heteroatomic substituent directly bonded to the allyl vinyl ether moiety.One of the few reported ex-amples of Johnson rearrangement with heteroatomic substitution (OCH 3)is the reaction of allylic alcohols with methyl methoxyorthoacetate that gives methyl R -methoxy-γ,δ-unsaturated esters in a process that occurs under acidic conditions (Scheme 13).183.4.Ireland −Claisen RearrangementIn 1972Ireland reported the rearrangement of allyl trimethylsilyl ketene acetals,19prepared by reaction of allylic ester enolates with trimethylsilyl chloride,to yield γ,δ-unsaturated carboxylic acids (Scheme 14).As compared with other reported rearrangements,this reaction proceeds under mild basic or neutral conditions.These conditions have allowed the preparation of polyfunctionalized structures,such as the vinylstan-nanes represented in Scheme 15.20This exampleprovides a method of functionalizing the newly formed double bond due to the high synthetic versa-tility of organotin compounds.3.5.Reformatsky −Claisen RearrangementWe have so far seen that allylic ester enolates rearrange quite easily.[3,3]Sigmatropic rearrange-Scheme10Scheme11Scheme12Scheme13Scheme14Scheme152942Chemical Reviews,2004,Vol.104,No.6Martı´n Castroment of zinc enolates,known as the Reformatsky -Claisen rearrangement,has also been reported.21These zinc enolates (10),generated by Reformatsky reaction of R -haloesters (11)with zinc dust,lead to the corresponding γ,δ-unsaturated zinc carboxylates (12)(Scheme 16)in good yields under neither acidic nor basic conditions.Similar reaction conditions,in the presence of trimethylsilyl chloride,allowed the synthesis of 2,2-difluoro-4-pentenoic acid starting from allyl chlorodi-fluoroacetate 22(Scheme 17).This silicon-inducedReformatsky -Claisen reaction did not occur in the absence of chlorotrimethylsilane.This indicates that the ketene acetal depicted in Scheme 17is most likely a reaction intermediate.3.6.Thio −Claisen RearrangementThermolysis of allyl phenyl sulfides (13)23leading to a [3,3]sigmatropic rearrangement contrasts with the classic Claisen rearrangement:it requires higher temperature to produce the corresponding thiols,intermediates which are not easily isolated and usually evolve into the corresponding diallyl deriva-tives due to a S N 2displacement by the intermediate thiolate on the starting sulfide (Scheme 18).24In contrast,the aliphatic version of the thio -Claisen rearrangement may proceed under milder conditions than those reported for oxygenated sub-strates (Scheme 19).25Nevertheless,the low applicability of this method-ology is a consequence of the instability of the prod-ucts.This prompted the development of conditions to trap and transform them into more stable com-pounds such as,for example,the hydrolysis of the intermediate thioaldehyde into the corresponding aldehyde (Scheme 20).263.7.Aza −Claisen RearrangementThe [3,3]sigmatropic rearrangement of N -allyl-N -arylamines,known as the aza -Claisen rearrange-ment (Scheme 21),27usually requires more drasticconditions than those required for the classic Claisen rearrangement of oxygenated substrates (this rear-rangement occurs at 200-350°C).In addition,it affords the corresponding anilines along with undes-ired byproducts.Similar energetic conditions are needed for the aliphatic aza -Claisen rearrangement to take place (Scheme 22).2a The thermal process requires highertemperatures than those needed for oxygen sub-strates.In a number of cases the reaction only evolves under Lewis-acid catalysis.3.8.Chelate Claisen RearrangementChelated enolates derived from amino acid esters undergo Claisen rearrangement upon standing at room temperature to produce γ,δ-unsaturated amino acids (Scheme 23).28Starting from E -allylic esters,syn products are obtained in a diastereoselective fashion.These reaction conditions are based on the fact that,in general,chelation sharply increases the thermal stability with no negative influence on the reactivity of those enolates.As a consequence of the rigid geometry of the enolate and the predictable geometry of the transition state,any transformationScheme20Scheme21Scheme22Scheme23Scheme16Scheme17Scheme18Scheme19Claisen Rearrangement over the Past Nine Decades Chemical Reviews,2004,Vol.104,No.62943will tend to take place with a very high stereoselec-tivity (see section 6.2.2).3.9.Diosphenol −Claisen RearrangementThis variety of Claisen rearrangement uses allyl ethers (14)derived from diosphenol,with an endocy-clic vinyl double bond,to give rise to a bond between a functionalized carbon moiety and a sterically hindered carbon which is part of a cyclic structure (Scheme 24).29The resulting bisketone usually tau-tomerizes into the ketoenol derivative.3.10.Metallo −Claisen RearrangementSeveral studies focused on the development of synthetic applications of gem -dimetallic compounds,30which were prepared by carbometalation of an al-kenyl organometallic magnesium,lithium,or alumi-num derivative with an allyl zinc bromide.The initially accepted pathway for the carbometalation consists first in the formation of an allyl vinyl zinc compound (15)(Scheme 25),which next undergoes a[3,3]rearrangement s a process known as the met-allo -Claisen rearrangement 30a s to afford the 1,1-bimetallic species 16.However,two mechanistic rationales,a metallo -ene reaction and a metallo -Claisen rearrangement,account for the resulting products as shown in Scheme 25.Density functional (B3LYP)studies on this reaction have demonstrated that the process is an endothermic Lewis-acid-as-sisted metallo -Claisen rearrangement with some character of metallo -ene reaction of the vinylmag-nesium species (MX n )MgCl in Scheme 25).31The high diastereoselectivity of the reaction has been explained by the short length of the forming C -C bond in the late transition state of the metallo -Claisen process.These organic gem -dimetallic compounds are able to react successively with two different electrophilesto produce various gem -difunctionalized structures 30b -d(Scheme 26).Several years later this methodology was expanded to the rearrangement of allyl allenyl derivatives to give access to gem -dimetalated dienes 30e,f (Scheme 27).As mentioned,recently a series of theoretical cal-culations has supported a mechanism in which the reaction of an allyl zinc bromide with a vinylmagne-sium bromide initially proceeds through a fast trans-metalation process to generate an allyl vinyl zinc intermediate A ,which undergoes a MgBr 2-assisted metallo -Claisen rearrangement through transition state B ,which generates the 1,1-dimetallic species C .The reaction product D results from a final oligomerization step 31(Scheme 28).An equilibriummixture of (E/Z )-allyl zinc bromide affords a single diastereomer resulting from reaction of the minor (Z )-allyl isomer.This result is explained by comparison of the relative energies of the diastereomeric transi-tion states since the pathway through the (Z )-isomer is favored over that evolving through the (E )reagent.3.11.Retro-Claisen RearrangementThe Claisen rearrangement,as in any other [3,3]sigmatropic rearrangement,takes place under ther-modynamic control.This reaction is irreversible toward the formation of the carbonyl compounds (Scheme 29)due to their higher thermodynamic stability.Scheme26Scheme27Scheme28Scheme29Scheme24Scheme252944Chemical Reviews,2004,Vol.104,No.6Martı´n CastroHowever,some structural features have been iden-tified as being responsible for inversion of the normal situation,favoring the transformation of the carbonyl compound into the vinyl ether.In this sense,both the presence of any substituent at a bridgehead position and vicinal quaternary carbons in the car-bonyl compound shifts the equilibrium toward the retro-Claisen isomer as a result of a relief in torsional strain (Scheme 30).32This effect is particularlymarked in the presence of a catalytic amount of Lewis acid (BF 3.OEt 2).This retro-Claisen process is general for a number of substrates containing contiguous quaternary centers whenever the R -carbonyl sub-stituent is not an electron-releasing group.A similar thermolability has been detected for vinylcyclopropane carboxaldehydes,which evolve into 2,5-dihydrooxepines by retro-Claisen rearrangement,as indicated in Scheme 31a.In this example thethermodynamic stability of the carbonyl group,which is favored at equilibrium,compensates for the un-stabilizing strain of the cyclopropane ring.33a The ready retro-Claisen reaction of this type of vinylcy-clopropanes is evidenced by a number of examples reported in the literature (see,for example,Scheme 31b 33b ).4.Mechanistic and Kinetic Aspects 4.1.General RemarksThe term “Claisen rearrangement”was originally applied to rearrangements of allyl aryl ethers to afford ortho-and occasionally para -substituted phe-nols.Afterward it expanded to analogue rearrange-ments of allyl vinyl ethers into unsaturated carbonyl compounds,which were classified as [3,3]sigmatropic rearrangements.34Initially a synchronic evolution for these reactions through aromatic transition states was accepted,35,36formed by a combination of σand πoverlap of 2p atomic orbitals of the carbon atoms of both allyl fragments.It was concluded that,out of the two feasible geometries for the transition state,the reaction proceeded through chairlike intermedi-ates (17)instead of boatlike intermediates (18)(Figure 1).37Both transition states (17and 18)arethe only ones corresponding to supra-supra processes and,therefore,allowed by Woodward -Hoffmann rules.34The intramolecular cyclic character of the rear-rangement is generally accepted.However,the re-search to understand the precise nature and the geometry of the transition state continues.A large number of theoretical calculations aiming to pre-dict the structures of the transition states involved in the Claisen rearrangement have been reported.38-41Most of them accept a concerted rearrangement through a chairlike transition state.However,re-cently Houk used quantum mechanical calculations to rationalize the stereoselectivity of the Ireland -Claisen rearrangement of cyclohexenyl silyl enol ethers from the chair or boat preferences in the transition state which derived from the substituents on the cyclohexenyl ring.38c In addition,there is no general agreement about the structure of this transi-tion state (Figure 2).42Over the decades several experimental studies based on kinetic isotopic effects have been reported in order to determine the geometry of the transition state of aliphatic and aromatic Claisen rearrange-ments.42-44However,this has not proved to be an easy task,and despite the numerous papers dealing with the Claisen rearrangement in different fields related to organic chemistry,there is no general agreement about such a geometry from theoretical predictions.The difficulty of describing such a ge-ometry still persists.4.2.Factors Affecting the Reaction RateThe most frequently reported Claisen rearrange-ment s thermal isomerization of allyl vinyl ethers s is a process that requires high temperatures and proceeds quite slowly at atmospheric pressure.To transform this reaction into a synthetically useful procedure,numerous attempts to find milder experi-mental conditions have been reported.The introduc-tion of different substituents in the carbon skeleton of the substrate as well as variations of the catalyst are worth mentioning.4.2.1.Influence of the SubstituentsIn the last 20years a considerable number of studies to determine the inductive or mesomeric effects of electron-withdrawing or electron-donating substituents located at different positions of the carbon skeleton have been mentioned.These effects are qualitatively described in Scheme 32.Carpenter studied the effect of the cyano group at different positions of allyl vinyl ethers.Such an effect was interpreted as basically electronic.45In the case of substrates substituted at positions C-2(k rel 111),C-4(k rel 270),and C-5(k rel 15.6),an acceleration of the rearrangement was detected,whereas substitu-Scheme30Scheme31Figure1.Figure 2.Claisen Rearrangement over the Past Nine Decades Chemical Reviews,2004,Vol.104,No.62945tion at C-1and C-6resulted in a decrease in the reaction rate.These observations were rationalized from Hu ¨ckel molecular orbital (HMO)theory,which allowed evaluation of the effect of a substituent in the transition state and in the ground state.45b The comparison of the difference of πenergy of HMO (∆E π)between the ground state and the transition state with the value of ∆E πfor the unsubstituted analogue compound allowed them to predict the sign and magnitude of the effect of the substituent in the activation enthalpy of the reaction.In this model the electron-withdrawing and -donating substituents are represented as carbocations and carbanions,respec-tively.From the delocalized model of the transition state,some qualitative predictions about the effects of the substituents in the Claisen rearrangement could be made.The main inconvenience of this model is that the cyano group is not only an electron-withdrawing group,but also a radical-stabilizing group,so that the acceleration resulting from the presence of a cyano group at C-2and C-4may not be a consequence of its electron-withdrawing character.To differentiate the inductive electron-withdrawing character and the result of a combination of inductive and mesomeric electron-withdrawing effects,the behavior of allyl vinyl ethers bearing a trifluorom-ethyl group at C-2and C-4was studied.46A CF 3group is an electron-withdrawing substituent with an inductive character but with no mesomeric one;it is not able to stabilize radicals.Hence,the Claisen rearrangement of allyl vinyl ethers bearing a CF 3group at C-2suffered an accelerating factor of 73in relation with the unsubstituted substrate,in com-parison with the value of k rel 111observed for cyano derivatives at C-2.45a In its turn,a CF 3group at C-4exerted no influence in the reaction rate.These results allowed Gajewski 46to suggest that the electron-withdrawing character of the substituent at C-4is not that responsible for the increase in the rate but its ability to stabilize radicals,which is reflected in the stabilization of the transition state.Similarly,quite recently it has been reported that a CF 3groupat C-1does not modify the reaction rate,whereas the effect of a fluorine atom at the same position will depend on the influence of an alkyl substituent R at C-2(Figure 3).47Different theoretical models predicting the effects of several substituents in the Claisen rearrangement rate have been proposed.The model suggested by Gajewski 48assumes that the structure of the transi-tion state adopts the features of the substrate or product depending on the exothermic properties of the reaction.In addition,it will have an associative or dissociative character according to the way that the substituents can stabilize such a character.Also,recently some theoretical calculations on the effects of cyano,amino,and trifluoromethyl substituents on the rate,whose results are coincident with those attained from experimental studies,have been re-ported.49The effect of alkoxy groups has been largely studied by Curran.50,51An electron-donating substituent (alkoxy group)at C-6sharply accelerates the Claisen rearrangement.50This effect seems to contradict Carpenter’s model,45which predicts a deceleration in the presence of a donating substituent at C-6,despite the loss of resonance energy from the ground state to the transition state.To compensate for this effect,the model proposes a πf σ*stabilization by a “vinylogous anomeric effect”of the O 3-C 4bond of the vinyl ether as responsible for the Claisen rearrange-ment acceleration by donating substituents at C-6.The transition state of the Claisen rearrangement (eq 2in Scheme 33)can be understood in a similar wayto the “double bond -no bond”resonance explaining the vinylogous anomeric effect (eq 1in Scheme 33).The process goes through an early transition state where the bond breaking is more advanced than the bond formation.45As can be seen,an oxygenated substituent at C-6must decrease the energy of the transition state s and,therefore,accelerate the reac-tion s as it makes the breaking of the weakened O 3-C 4bond easier.A similar acceleration was detected with the pres-ence of an alkoxy group at C-4.In addition,the rates of the rearrangements of 4-and 6-alkoxyallyl enol ethers were quite sensitive to the solvent polarity and considerably increased in hydrogen bonding sol-vents 51s it could not be detected from unsubstituted substrates.These results were attributed to anScheme32Figure 3.Scheme332946Chemical Reviews,2004,Vol.104,No.6Martı´n Castro。
有机化学重排反应-总结
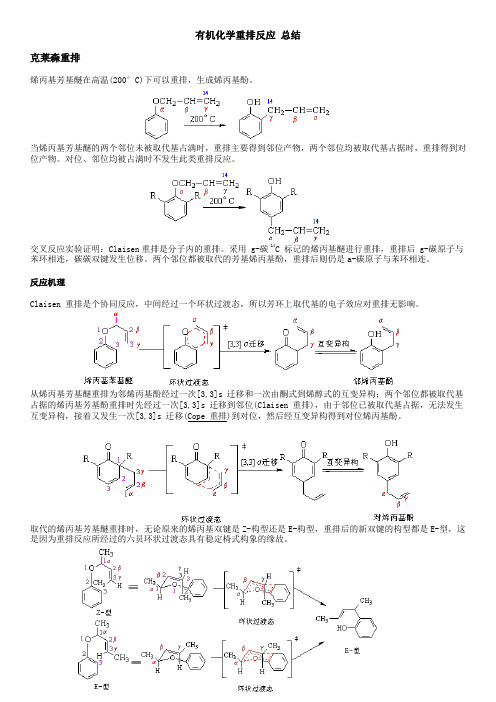
有机化学重排反应总结克莱森重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚。
当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要得到邻位产物,两个邻位均被取代基占据时,重排得到对位产物。
对位、邻位均被占满时不发生此类重排反应。
交叉反应实验证明:Claisen重排是分子内的重排。
采用 g-碳 14C 标记的烯丙基醚进行重排,重排后 g-碳原子与苯环相连,碳碳双键发生位移。
两个邻位都被取代的芳基烯丙基酚,重排后则仍是a-碳原子与苯环相连。
反应机理Claisen 重排是个协同反应,中间经过一个环状过渡态,所以芳环上取代基的电子效应对重排无影响。
从烯丙基芳基醚重排为邻烯丙基酚经过一次[3,3]s 迁移和一次由酮式到烯醇式的互变异构;两个邻位都被取代基占据的烯丙基芳基酚重排时先经过一次[3,3]s 迁移到邻位(Claisen 重排),由于邻位已被取代基占据,无法发生互变异构,接着又发生一次[3,3]s 迁移(Cope 重排)到对位,然后经互变异构得到对位烯丙基酚。
取代的烯丙基芳基醚重排时,无论原来的烯丙基双键是Z-构型还是E-构型,重排后的新双键的构型都是E-型,这是因为重排反应所经过的六员环状过渡态具有稳定椅式构象的缘故。
Claisen 重排具有普遍性,在醚类化合物中,如果存在烯丙氧基与碳碳相连的结构,就有可能发生Claisen 重排。
2.Beckmann贝克曼重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:3.Bamberger,E.重排苯基羟胺〔N-羟基苯胺〕和稀硫酸一起加热发生重排成对-氨基苯酚:在H2SO4-C2H5OH(或CH3OH)中重排生成对-乙氧基〔或甲氧基〕苯胺:其他芳基羟胺,它的环上的o-p位上未被取代者会起类似的重排。
claisen重排应用实例

claisen重排应用实例
克莱森重排(Claisen rearrangement)是一种重要的有机化学反应,它可以将一个醇或酚转化为相应的酯。
这个反应在有机合成
中具有广泛的应用,下面给出一个典型的克莱森重排应用实例:
实例:合成乙酰乙酸异丙酯
步骤1:合成乙酰乙酸乙酯
首先,将乙醇和乙酸酐(酸酐是酸的酯化合物)加入反应瓶中,并加入一定量的碱催化剂,如氢氧化钠(NaOH)。
反应瓶通常使用
玻璃瓶,配有反应器和冷凝器。
乙醇 + 乙酸酐→ 乙酸乙酯 + 乙酸
此反应是一个酯化反应,通过酸催化生成酯。
反应进行时,反
应瓶中的温度应保持在适宜的范围内,通常在60-80摄氏度之间。
步骤2:克莱森重排反应
将合成的乙酸乙酯加入反应瓶中,并加入一定量的碱催化剂,
如氢氧化钠(NaOH)。
反应瓶中的温度应保持在适宜的范围内,通
常在100-150摄氏度之间。
乙酸乙酯→ 乙酸异丙酯
此反应是克莱森重排反应,通过碱催化使酯分子内部的酯基迁移,生成相应的酯。
步骤3:提取产物
反应结束后,将反应瓶中的产物进行提取。
通常使用有机溶剂,如乙醚或氯仿,将产物从反应混合物中分离出来。
步骤4:纯化产物
提取得到的产物可能还含有杂质,需要进行纯化。
可以使用色
谱层析等技术将产物纯化。
通过以上步骤,可以成功合成乙酰乙酸异丙酯。
这个反应实例
展示了克莱森重排的应用,通过酯化和重排反应,可以将乙醇和乙
酸酐转化为乙酰乙酸异丙酯。
克莱森重排反应机理

克莱森重排反应是有机化学中一种重要的反应类型,常用于合成芳香化合物。
本文将从反应机理的角度对克莱森重排反应进行详细介绍。
克莱森重排反应是指通过醛或酮与二酮或二醇在碱性条件下反应,生成α,β-不饱和酮的反应。
该反应通常在碱性条件下进行,碱的作用是使醛或酮发生亲核加成,生成一个稳定的醇醚中间体。
这个中间体经过质子转移和脱水反应,形成α,β-不饱和酮产物。
克莱森重排反应的机理可以分为三步。
首先,醛或酮与碱反应生成醇醚中间体。
这个中间体是通过醛或酮的羰基碳上的亲核加成形成的。
其次,醇醚中间体发生质子转移反应,质子从羰基碳上转移到相邻的碳上,形成一个稳定的碳阳离子中间体。
最后,碳阳离子中间体通过脱水反应,失去一个水分子,生成α,β-不饱和酮产物。
克莱森重排反应的机理中,质子转移是一个关键步骤。
质子转移的位置取决于碱的性质以及反应条件。
通常情况下,质子转移到离羰基碳最近的一个碳上,形成稳定的碳阳离子。
然而,在特殊情况下,质子转移也可能发生到离羰基碳更远的位置上。
克莱森重排反应的应用非常广泛。
它可以用于合成芳香化合物,尤其是那些具有α,β-不饱和酮结构的化合物。
这类化合物在药物合成和天然产物合成中具有重要的地位。
此外,克莱森重排反应还可以用于合成含有杂环结构的化合物,如吡咯、噻吩等。
总之,克莱森重排反应是一种重要的有机化学反应,通过醛或酮与二酮或二醇在碱性条件下反应,生成α,β-不饱和酮。
该反应的机理包括醛或酮的亲核加成、质子转移和脱水反应等步骤。
克莱森重排反应在有机合成中有广泛的应用,特别是在合成芳香化合物和含有杂环结构的化合物方面。
克莱森重排反应

克莱森重排反应(Claisen重排反应)的最初形式是一个烯丙基苯基醚在高温(> 200°C)下发生的一个重排反应,产物是邻位烯丙基苯酚。
反应的机理是σ[3,3]重排(是史上第一个发现的σ[3,3]重排反应),产物4-烯酮因芳香性的需要互变异构为酚。
这个反应的特点是高度的区域选择性,产物大部分是邻位的。
与弗里斯重排的性质很相似。
而当苯环的两个邻位都被“堵”住的时候,反应产物是对位烯丙基取代物。
这是因为中间产物发生了一个科普重排反应所致——“分子自有其道(molecules have a way of hanging on)”。
审视整个过程可以看到:克莱森重排的驱动力是生成热力学上最稳定的取代度最大的“烯烃”。
克莱森重排起初是在芳香化合物中发现的(1912年),这与当时(20世纪初期)合成化学家“玩”的范围局限在芳香烃上有关。
到后来发现该反应可以拓展到非芳香化合物,而这种拓展非常重要,因为克莱森重排反应立刻变成了合成上一个非常有用的反应:反应生成了一个新的碳碳键,得到一个4-烯羰基化合物,而烯键可以继续往下做衍生,得到其他的化合物。
而现代有机合成在克莱森反应的启发下催生出众多“变体”:贝勒斯(Bellus)变体:埃申莫瑟(Eschenmoser)变体:艾兰德(Ireland)变体:强生(Johnson)变体:[编辑]天然界的存在在植物代谢的莽草酸途径中从分支酸到预苯酸的转换步骤就是一个克莱森重排;该反应受分支酸歧化酶的催化。
预苯酸是一个重要的前体化合物,生物体内含苯环的天然化合物有一大半是由预苯酸转换过来的。
嚬哪醇重排(英:pinacol rearrangement)是一个邻二醇在酸催化下脱水并发生取代基重排生成羰基化合物的反应。
[1][2]。
这一类反应由于嚬哪醇(2,3-二甲基-2,3-丁二醇)转换为嚬哪酮(3,3-二甲基-2-丁酮)的反应最具代表性,因而得名。
反应的关键步骤是一个碳正离子的1,2-重排。
克莱森重排机理

克莱森重排机理克莱森重排是指在存在乙酰丙酮(或酮类)和硫脲(或脲类)等分子间的反应中,分子间发生重排反应,生成异构体的化学过程。
该反应在有机化学合成中有广泛的应用,具有一定的机理特点。
一、反应机理克莱森重排的反应机理包括以下几个步骤:1.硫脲与酮类分子发生亲核加成,形成价态复杂的中间体;2.中间体负离子发生轴向异构,形成中间体B。
此时,碳负离子在α位置得到了稳定化;3.负离子中间体B发生质子转移,并发生环型闭合,生成异构体。
此时,杂原子硫原子转化为环内原子性质,生成六元环的化合物;4.反应的最终产物是异构体的混合物,难以区分哪个成分是主要的产物。
二、反应条件克莱森重排作为一种常用的有机化学反应,其反应条件具有一定的特点,通常需要满足以下条件:1.反应物要求有一定的亲电性和亲核性,包括酮类和硫脲(或脲类)化合物;2.反应需要在适量的碱性条件下进行,以促进负离子中间体的形成;3.反应需要在适宜的温度下进行,不宜过高或过低,一般在室温至70℃之间;4.溶剂选择要合适,一般选择乙醇、二甲基甲酰胺等极性溶剂;5.该反应具有缩合反应的性质,易于生成大分子化合物。
三、应用克莱森重排在有机化学领域中具有广泛的应用,常用于:1.生产有机化合物的制备,例如生产杀菌剂、农药、抗生素等产品;2.新药研发领域中的化学反应研究;3.有机合成化学研究领域中,用于生成环状或多环状的复杂化合物,为后续有机合成研究提供组成部分;4.生物化学领域,用于设计合成有机化合物,以研究其生物活性等性质。
总之,克莱森重排反应是一种常用的有机反应过程,具有一定的机理特征和应用价值。
在化学研究领域,在其反应条件和应用范围等方面进行深入了解和研究,对于有机化学领域的发展和推动具有重要意义。
克莱森重排

O O
Ph
O
O
Ph heat
OH
OH
Ph
+ NH2
+ NH2
自1950年以来,化学家们通过标记技术,立体化学探针,动力学分析,分子间和分 子内的交叉实验等方法探究该反应的机理。对于烯丙基苯基醚型的克莱森重排,还 检测和直接研究了反应中间体环己二烯酮。通常克莱森重排机理有两种表示方法: 用双箭头表示电子转移反应式或者用单箭头表示单电子转移反应式
有时候重排反应会发生在烯丙基的β-碳原子上,并形成非正常的克莱森重排反应产物。 机理研究表明,非正常的重排产物是通过正常的邻位重排产物生成的。如下图所示: 正常的邻位重排产物中的酚羟基上的氢原子首先转移到烯丙基的末端碳原子上,然后 形成由环丙烷和环己二烯酮形成的螺环中间体。当环己二烯酮的氧原子从环丙烷上取代 基的碳原子上夺取一个氢原子后,环丙烷开环重排到烯丙基β-碳原子上。
CH3C(OMe)3 EtCO2H,140C,3h 83%
OEt O O H O O H
O MeOOC
O OMe
OMe H O O H
O
MeOOC 3(MeO)C COOMe O
xylene,160C,1h
H O HOBiblioteka H O59%O MeO2C CO2Me H O H O O
CO2Me
O
O
CO2H
CH3 O heat H C H 2 =C H CH3 (E ,Z )o r(Z ,E) (Z ,Z )or (E ,E) 3% 9 7 .8% 97% 2 .2% CHO + H H H CH3 CHO CH=CH2 CH3
(E,E)-丙烯基巴豆基醚在椅式过渡态中,两个甲基都处于椅式的平伏键上。由于 这种构象能量最低而最容易发生重排,所以生成主要产物。而在船式六元环状过渡态 中,两个甲基则都处于准平伏键上。由于船底的两组碳原子为重叠式构象,能量相对 较高而较难发生重排,因此生成次要产物。
克莱森缩合反应ClaisenCondensation

第7页/共10页
应用举例
1.乙酰乙酸乙酯的合成 两分子乙酸乙酯可以通过该反应缩合成为3羰基丁酸乙酯(通常称为乙酰乙酸乙酯), 该化合物是有机合成的重要中间体。
Claisen是一个很巧的和富于创造力的 化学家。当你在图书馆查找有机化学方面 的文献时会将会碰到Claisen所做的一些工 作。他的成就包括羰基化合物的酰化,烯 丙基重排(Claisen重排)),肉桂酸 (PhCH=CHCOOH)的制备,吡唑(邻二 氮杂茂)的合成,异哑唑衍生物的合成和 乙酰乙酸乙酯的制备。
第4页/共10页
几种特殊的反应
2.位阻很大的格利雅试剂不能与羰基加成, 但可以夺取活性氢,也可以用来使羧酸酯 变成烯醇盐:
第5页/共10页
几种特殊的反应
3.两种不同的酯也能发生酯缩合, 理论上可得到四种不同的产物, 称为混合酯缩合,在制备上没 有太大意义。如果其中一个酯 分子中既无α-氢原子,而且 烷氧羰基又比较活泼时,则仅 生成一种缩合产物。如苯甲酸 酯、甲酸酯、草酸酯、碳酸酯 等,与其它含α-氢原子的酯 反应时,都只生成一种缩合产 物。
第10页/共10页
第1页/共10页
基本定义
克莱森缩合反应(Claisen Condensation):
两分子羧酸酯在强碱(如乙醇 钠)催化下,失去一分子醇而缩合 为一分子β-羰基羧酸酯的反应。参 与反应的两个酯分子不必相同,但 其中一个必须在酰基的α-碳上连有 至少一个氢原子。简单的说,该反 应是一个酯分子的酰基对另一酯分 子的酰基α-碳原子进行的酰化反应。
第8页/共10页
应用举例
2.乙酰丙酮的合成 在乙醇钠的催化下,一分子乙酸乙酯和一 分子丙酮可缩合为2,4-戊二酮(通常称为 乙酰丙酮)。乙酰丙酮的共轭碱在配位化 学中是重要的配体。
Claisen重排

凡是含有两个双键和六个原子,且一个双键和杂 原子X(X为O、S、N等)有共轭关系的化合物 所发生的重排反应,统称为Claisen重排。
一般形式:
Claisen重排最初形式
烯丙基苯基醚在高温(> 200°C)下发生的一个重 排反应这,个产反物应是的邻特位点烯是丙高基度苯的酚区。域反选应择的性机,理是 σ[3,3]产重物排大(部是分史是上邻第位一的个。发现的σ[3,3]重排反应), 产物4-烯酮因芳香性的需要互变异构为酚。
脂肪族Claisen反应机理
Mechanism of the Claisen Rearrangement
The aliphatic Claisen Rearrangement is a [3,3]-sigmatropic rearrangement in which an allyl vinyl ether is converted thermally to an unsaturated carbonyl compound
脂肪族Claisen反应机理
The reaction proceeds preferably via a chair transition state. Chiral, enantiomerically enriched starting materials give products of high optical purity
Claisen重排反应应用
邻丁子香酚
Claisen重排反应应用
手性氮氧-镍(II)配合物催化的不对称炔丙基和烯 丙基的克莱森重排反应,成功构建一系列联烯基和 烯丙基取代的季碳中心
Cope重排
克莱森重排
4.5水相中的克莱森重排反应 水相中的克莱森重排反应 使用水溶剂可以加速克莱森重排反应,并且能够在温和的条件下进行
O PhMe,100C,95h 12% (CH2)5CO2R (CH2)5 CO2R CHO
4.6高压克莱森重排反应 高压克莱森重排反应 烯丙基醚(对甲基苯基醚)在不同的温度和溶剂中发生克莱森重排的速率常数都会随 压力的增大而增大。间甲氧基醚和烯丙基乙烯基醚也有类似的现象。
OH OH heat + + Ph
O O
Ph
O
O
Ph heat
OH
OH
Ph
+ NH2
+ NH2
自1950年以来,化学家们通过标记技术,立体化学探针,动力学分析,分子间和分 子内的交叉实验等方法探究该反应的机理。对于烯丙基苯基醚型的克莱森重排,还 检测和直接研究了反应中间体环己二烯酮。通常克莱森重排机理有两种表示方法: 用双箭头表示电子转移反应式或者用单箭头表示单电子转移反应式
CH3 O heat H C H 2 =C H CH3 (E ,Z )o r(Z ,E) (Z ,Z )or (E ,E) 3% 9 7 .8% 97% 2 .2% CHO + H H H CH3 CHO CH=CH2 CH3
(E,E)-丙烯基巴豆基醚在椅式过渡态中,两个甲基都处于椅式的平伏键上。由于 这种构象能量最低而最容易发生重排,所以生成主要产物。而在船式六元环状过渡态 中,两个甲基则都处于准平伏键上。由于船底的两组碳原子为重叠式构象,能量相对 较高而较难发生重排,因此生成次要产物。
由于克莱森重排反应可以方便的通过椅式构象过渡态控制产 物的立体化学。因此克莱森重排反应已经广泛应用于天然产 物的全合成中。在含有季碳的萜类天然产物中,都是使用脂 肪克莱森重排反应作为关键步骤实现的。
有机化学重排反应 总结

有机化学重排反应总结克莱森重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚。
当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要得到邻位产物,两个邻位均被取代基占据时,重排得到对位产物。
对位、邻位均被占满时不发生此类重排反应。
交叉反应实验证明:Claisen重排是分子内的重排。
采用 g-碳 14C 标记的烯丙基醚进行重排,重排后 g-碳原子与苯环相连,碳碳双键发生位移。
两个邻位都被取代的芳基烯丙基酚,重排后则仍是a-碳原子与苯环相连。
反应机理Claisen 重排是个协同反应,中间经过一个环状过渡态,所以芳环上取代基的电子效应对重排无影响。
从烯丙基芳基醚重排为邻烯丙基酚经过一次[3,3]s 迁移和一次由酮式到烯醇式的互变异构;两个邻位都被取代基占据的烯丙基芳基酚重排时先经过一次[3,3]s 迁移到邻位(Claisen 重排),由于邻位已被取代基占据,无法发生互变异构,接着又发生一次[3,3]s 迁移(Cope 重排)到对位,然后经互变异构得到对位烯丙基酚。
取代的烯丙基芳基醚重排时,无论原来的烯丙基双键是Z-构型还是E-构型,重排后的新双键的构型都是E-型,这是因为重排反应所经过的六员环状过渡态具有稳定椅式构象的缘故。
反应实例Claisen 重排具有普遍性,在醚类化合物中,如果存在烯丙氧基与碳碳相连的结构,就有可能发生Claisen 重排。
贝克曼重排肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例,E.重排苯基羟胺(N-羟基苯胺)和稀硫酸一起加热发生重排成对-氨基苯酚:在H2SO4-C2H5OH(或CH3OH)中重排生成对-乙氧基(或甲氧基)苯胺:其他芳基羟胺,它的环上的o-p位上未被取代者会起类似的重排。
克莱森重排反应
克莱森重排反应(Claisen重排反应)的最初形式是一个烯丙基苯基醚在高温(> 200°C)下发生的一个重排反应,产物是邻位烯丙基苯酚。
反应的机理是σ[3,3]重排(是史上第一个发现的σ[3,3]重排反应),产物4-烯酮因芳香性的需要互变异构为酚。
这个反应的特点是高度的区域选择性,产物大部分是邻位的。
与弗里斯重排的性质很相似。
而当苯环的两个邻位都被“堵”住的时候,反应产物是对位烯丙基取代物。
这是因为中间产物发生了一个科普重排反应所致——“分子自有其道(molecules have a way of hanging on)”。
审视整个过程可以看到:克莱森重排的驱动力是生成热力学上最稳定的取代度最大的“烯烃”。
克莱森重排起初是在芳香化合物中发现的(1912年),这与当时(20世纪初期)合成化学家“玩”的范围局限在芳香烃上有关。
到后来发现该反应可以拓展到非芳香化合物,而这种拓展非常重要,因为克莱森重排反应立刻变成了合成上一个非常有用的反应:反应生成了一个新的碳碳键,得到一个4-烯羰基化合物,而烯键可以继续往下做衍生,得到其他的化合物。
而现代有机合成在克莱森反应的启发下催生出众多“变体”:贝勒斯(Bellus)变体:埃申莫瑟(Eschenmoser)变体:艾兰德(Ireland)变体:强生(Johnson)变体:[编辑]天然界的存在在植物代谢的莽草酸途径中从分支酸到预苯酸的转换步骤就是一个克莱森重排;该反应受分支酸歧化酶的催化。
预苯酸是一个重要的前体化合物,生物体内含苯环的天然化合物有一大半是由预苯酸转换过来的。
嚬哪醇重排(英:pinacol rearrangement)是一个邻二醇在酸催化下脱水并发生取代基重排生成羰基化合物的反应。
[1][2]。
这一类反应由于嚬哪醇(2,3-二甲基-2,3-丁二醇)转换为嚬哪酮(3,3-二甲基-2-丁酮)的反应最具代表性,因而得名。
反应的关键步骤是一个碳正离子的1,2-重排。
Claisen重排反应

Claisen重排反应Claisen重排反应是由Rainer Ludwig Claisen在1912年发现,是第一个被发现的[3,3]-σ迁移重排。
此反应是强力生成碳碳键的反应,加热烯丙基烯基醚通过[3,3]-σ迁移重排生成γ,δ-不饱和羰基化合物的反应。
另外对位-Claisen重排, Belluš–Claisen r重排; Corey–Claisen 反应, Eschenmoser–Claisen重排, Ireland–Claisen反应, Kazmaier–Claisen反应, Saucy–Claisen反应; orthoester Johnson–Claisen反应和Carroll重排都属于这个系列的反应。
Claisen重排反应是协同反应。
反应机理Claisen重排反应属于放热反应 (大约84 kJ/mol),属于协同周环反应,遵循Woodward-Hoffmann规则的同侧反应路径。
此反应的溶剂效应明显,大极性溶剂会加速反应,质子化溶剂反应速率较快,如乙醇/水体系溶剂将近是环丁砜溶剂的10倍。
三价有机铝试剂,如三甲基铝可以加速反应。
反应实例芳基Claisen重排Bellus-Claisen重排Eschenmoser-Claisen重排Ireland-Claise重排Johnson-Claisen重排参考文献1. Claisen, L. Ber. 1912, 45, 3157 -3166.2. Rhoads, S. J.; Raulins, N. R. Org. React. 1975, 22, 1- 252. (Review).3. Wipf, P. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.;Pergamon, 1991, Vol. 5, 827- 873. (Review).4. Ganem, B. Angew. Chem. Int. Ed. 1996, 35, 937- 945. (Review).5. Ito, H.; Taguchi, T. Chem. Soc. Rev. 1999, 28, 43- 50. (Review).6. Castro, A. M. M. Chem. Rev. 2004, 104, 2939 -3002. (Review).7. Jürs, S.; Thiem, J. Tetrahedron: Asymmetry 2005, 16, 1631- 1638.8. Vyvyan, J. R.; Oaksmith, J. M.; Parks, B. W.; Peterson, E. M. Tetrahedron Lett. 2005,46, 2457 2460.9. Nelson, S. G.; Wang, K. J. Am. Chem. Soc. 2006, 128, 4232–4233.10. Körner, M.; Hiersemann, M. Org. Lett. 2007, 9, 4979–4982.11. Uyeda, C.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 9228–9229.12. Williams, D. R.; Nag, P. P. Claisen and Related Rearrangements. In Name Reactions for Homologations-Part II; Li, J. J., Ed.; Wiley: Hoboken, NJ, 2009, pp 33 43. (Review).13. Alwarsh, S.; Ayinuola, K.; Dormi, S. S.; McIntosh, M. C. Org. Lett. 2013, 15, 3–5.编译自:J.J. Li, Name Reactions: A Collection of DetailedMechanisms and Synthetic Applications, Claisen rearrangements,page 140-141.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
同)的则给出成键轨道,两个原子轨道的对称性不
同(位相不同)的则给出反键轨道。
原子轨道 S 轨道 P 轨道
图形
对称
不对称
第一节
周环反应的理论>二、周环反应的理论
分子轨道对称守恒原理中心内容及内函: 化学反应是分子轨道重新组合的过程,分 子轨道的对称性控制化学反应的进程,在一个
协同反应中,分子轨道对称性守恒。(即在一
反应物→中间体→产物 周环反应: 反应物→产物
第 一节
周环反应的理论
一、周环反应 二、周环反应的理论
第一节
周环反应的理论>一、周环反应
周环反应的特征: (1)多中心的一步反应,反应进行时键的断裂和 生成是同时进行的(协同反应)。
(2)反应进行的动力是加热或光照。不受溶剂 极性影响,不被酸碱所催化,不受任何引发剂的引发。
,用LUMO表示。
HOMO、LUMO统称为前线轨道,处在前线轨道上的电 子称为前线电子。 有的共轭体系中含有奇数个电子,它的已占有电子 的能级最高的轨道中只有一个电子,这样的轨道称为单
占轨道,用SOMO表示,单占轨道既是HOMO,又是LUMO。
第一节
周环反应的理论>二、周环反应的理论
ψ4
E4
ψ
3
E3 E
曾昭琼《有机化学》第三版CAI教学配套课件
有机化学
主讲:谢启明 教授
第十七章
周环反应
§17~1 周环反应的理论
§17~2 电环化反应
§17~3 σ- 键迁移反应
§17~4 环加成反应
第一节
周环反应的理论>一、周环反应
周环反应:反应中无中间体生成,而是通过形 成过渡态一步完成的多中心反应。
离子型或游离基型反应:
电环化反应是分子内的周环反应,电环化反应的 成键过程取决于反应物中开链异构物的HOMO轨道的对
称性。
第二节 电环化反应>一、含4n个π电子体系的电环化 以丁二烯为例讨论——丁二烯电环化成环丁烯时, 要求: 1、C1—C2,C3—C4沿着各自的键轴旋转,使C1和C4 的 轨道结合形成一个新的σ -键。
第一节
周环反应的理论>二、周环反应的理论
1、σ-键的形成 当两个原子轨道沿着键轴方向对称重叠时,可形 成两个σ -键的分子轨道。对称性相同的原子轨道形 成σ -成键轨道,对称性不同的原子轨道形成σ *成键 轨道。
第一节
周环反应的理论>二、周环反应的理论
第一节
周环反应的理论>二、周环反应的理论
第一节
第一节
周环反应的理论>一、周环反应
(3)反应有突出的立体选择性,生成空间定向 产物。
R R
hυ
R R R = -COOCH 3 R
R
第一节
周环反应的理论>二、周环反应的理论
(一)轨道和成键
反键轨道 X1 成键轨道 X2 原子轨道
第一节
周环反应的理论>二、周环反应的理论
(二)分子轨道对称守恒原理 原子轨道组合成分子轨道时,遵守轨道对称守 恒原理。即当两个原子轨道的对称性相同(位相相
丁二烯在基态(加热)环化时,起反应的 前线轨道HOMO是ψ 2:
第二节 电环化反应>一、含4n个π电子体系的电环化 所以4nπ 丁二烯在基态(加热)环化时,顺 旋允许,对旋禁阻。
对旋 禁阻的
第二节 电环化反应>一、含4n个π电子体系的电环化 顺旋允许
第二节 电环化反应>一、含4n个π电子体系的电环化
有电子给予体的性质,而LUMO则对电子的亲和力较强,
具有电子接受体的性质,这两种轨道最易互相作用,在 化学反应过程中起着极其重要作用。
第一节
周环反应的理论>二、周环反应的理论
电环化反应
环加成反应
迁移反应
第二节
电环化反应
一、含4n个π电子体系的电环化 二、4n+2个π电子体系的电环化
第二节 电环化反应 电环化反应是在光或热的条件下,共轭多烯烃的两 端环化成环烯烃和其逆反应。
ψ2
E2
ψ1
E1 基态 丁二烯的分子轨道图 激发态
第一节
周环反应的理论>二、周环反应的理论
第一节
周环反应的理论>二、周环反应的理论
第一节
周环反应的理论>二、周环反应的理论
第一节
周环反应的理论>二、周环反应的理论
2. 前线轨道理论的中心思想 前线轨道理论认为:分子中有类似于单个原子的“ 价电子”的电子存在,分子的价电子就是前线电子,因 此在分子之间的化学反应过程中,最先作用的分子轨道 是前线轨道,起关键作用的电子是前线电子。 这是因为分子的HOMO对其电子的束缚较为松弛,具2 源自 3 4hv 对旋1
2
3
4
第二节 电环化反应>一、含4n个π电子体系的电环化 对旋允许
第二节 电环化反应>一、含4n个π电子体系的电环化 顺旋是禁阻
第二节 电环化反应>一、含4n个π电子体系的电环化 其他含有π 电子数为4n的共轭多烯烃体系的电 环化反应的方式也基本相同。例如:
第二节 电环化反应>一、含4n个π电子体系的电环化 对旋禁阻
第二节 电环化反应>一、含4n个π电子体系的电环化
丁二烯在激发态(光照)环化时,起反应 的前线轨道HOMO是ψ 3
第二节 电环化反应>一、含4n个π电子体系的电环化 所以4n+2π 丁二烯在激发态(光照)环化时, 对旋允许,顺旋是禁阻的。
周环反应的理论>二、周环反应的理论
2.π-键的形成 当两个P轨道侧面重叠时,可形成两个π 分子轨 道。对称性相同的P轨道形成成键π 轨道。对称性不 同的P轨道形成反键π *轨道。
第一节
周环反应的理论>二、周环反应的理论
1. 前线轨道和前线电子 已占有电子的能级最高的轨道称为最高占有轨道, 用HOMO表示。 未占有电子的能级最低的轨道称为最低未占有轨道
个协同反应中,由原料到产物,轨道的对称性 始终保持不变)。因为只有这样,才能用最低 的能量形成反应中的过渡态。
第一节
周环反应的理论>二、周环反应的理论
分子轨道对称守恒原理有三种理论解释:
前线轨道理论;
能量相关理论;
休克尔-莫比乌斯结构理论(芳香过渡态理论)。
这几种理论各自从不同的角度讨论轨道的对称性。 其中前线轨道理论最为简明,易于掌握。
2、旋转的方式有两种,顺旋和对旋。
第二节 电环化反应>一、含4n个π电子体系的电环化 3、反应是顺旋还是对旋,取决于分子是基态 还是激发态时的HOMO轨道的对称性。 4
(LUMO)
(HOMO)
3
2 1
(HOMO)
Ground state
Excited state
第二节 电环化反应>一、含4n个π电子体系的电环化