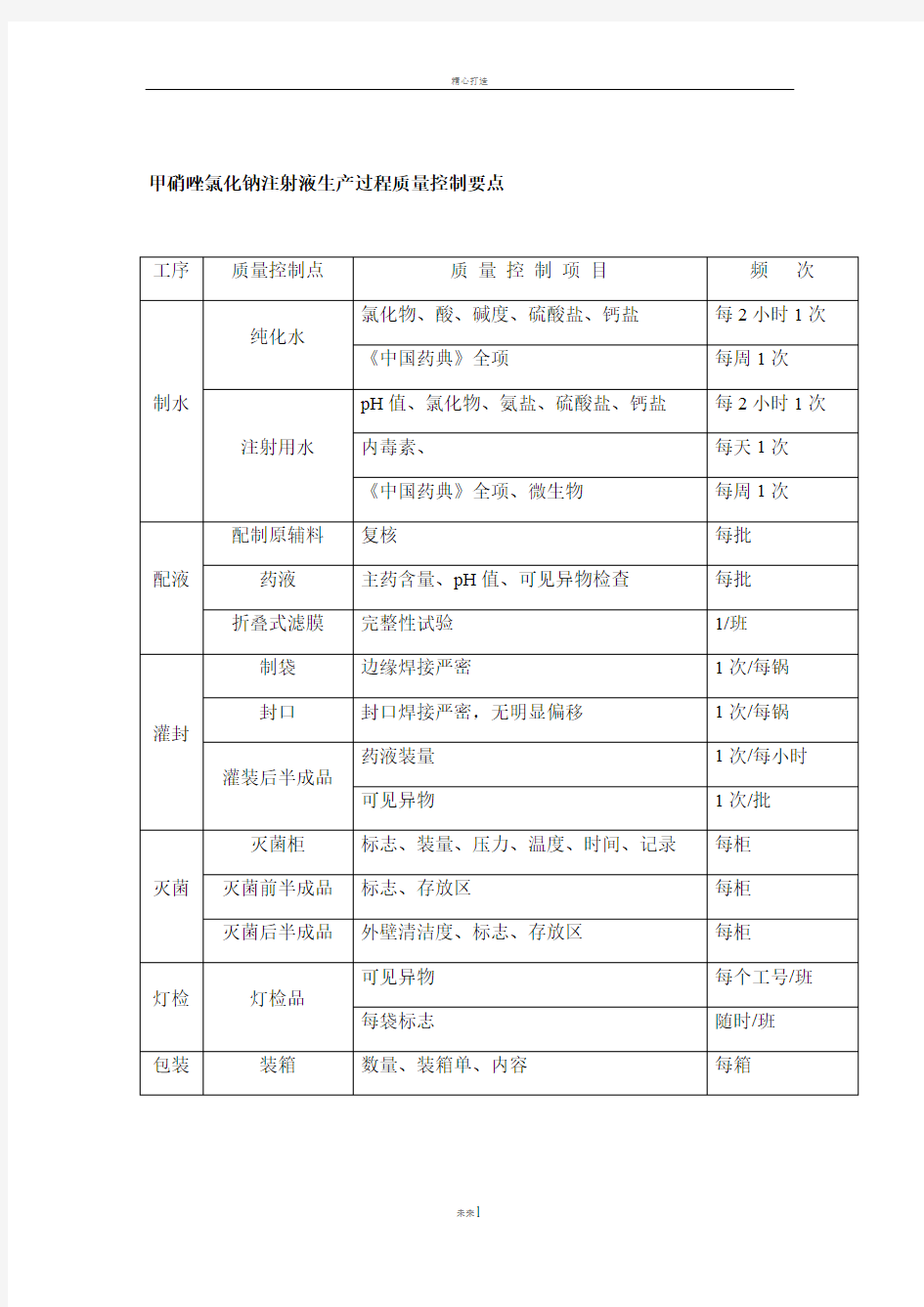
甲硝唑氯化钠注射液生产过程质量控制要点
精心打造甲硝唑氯化钠注射液生产过程质量控制要点
未来1
药品生产质量管理制度
药品生产质量管理 制度 1 2020年4月19日
第一章总则 第一条为规范药品生产质量管理,根据<中华人药品生产质量管理规范( 修订) 民共和国药品管理法>、<中华人民共和国药品管理法(卫生部令第79号) 实施条例>,制定本规范。 第二条企业应当建立药品质量管理体系。该体<药品生产质量管理规范( 修订)>已于 10月19日 系应当涵盖影响药品质量的所有因素,包括确保药品质经卫生部部务会议审议经过,现予以发布,自 3月1日 量符合预定用途的有组织、有计划的全部活动。 起施行。 第三条本规范作为质量管理体系的一部分,是药 品生产管理和质量控制的基本要求,旨在最大限度地降 部长陈竺 低药品生产过程中污染、交叉污染以及混淆、差错等 风险,确保持续稳定地生产出符合预定用途和注册要求 二○一一年一月十七 的药品。 日 第四条企业应当严格执行本规范,坚持诚实守信, 禁止任何虚假、欺骗行为。 第二章质量管理 1 2020年4月19日
2 2020年4月19日 第一节 原 则 第五条 企业应当建立符合药品质量管理要求的质量目标,将药品注册的有关安全、有效和质量可控的所有要求,系统地贯彻到药品生产、控制及产品放行、贮存、发运的全过程中,确保所生产的药品符合预定用途和注册要求。 第六条 企业高层管理人员应当确保实现既定的质量目标,不同层次的人员以及供应商、经销商应当共同参与并承担各自的责任。 第七条 企业应当配备足够的、符合要求的人员、厂房、设施和设备,为实现质量目标提供必要的条件。 第二节 质量保证 第八条 质量保证是质量管理体系的一部分。企业必须建立质量保证系统,同时建立完整的文件体系,以保证系统有效运行。 第九条 质量保证系统应当确保: (一)药品的设计与研发体现本规范的要求; (二)生产管理和质量控制活动符合本规范的要求; (三)管理职责明确; (四)采购和使用的原辅料和包装材料正确无误; (五)中间产品得到有效控制; (六)确认、验证的实施; (七)严格按照规程进行生产、检查、检验和复核; (八)每批产品经质量受权人批准后方可放行; (九)在贮存、发运和随后的各种操作过程中有保证药品质量的适当措施; (十)按照自检操作规程,定期检查评估质量保证系统的有效性和适用性。 第十条 药品生产质量管理的基本要求: (一)制定生产工艺,系统地回顾并证明其可持续稳
9105 多晶型药品的质量控制技术与方法指导原则
9105
多晶型药品的质量控制技术与方法指导原则
固体药物及其制剂中存在多晶型现象时,应使用“优势药物晶型物质状态”作为药物原 料及其制剂晶型,以保证药品临床有效性、安全性与质量可控性。 当固体药品存在多晶型现象且不同晶型物质状态可影响药品的有效性、安全性与药品质 量时,应对固体制剂、半固体、悬浮剂等制剂种类中的原料药晶型物质状态进行定性、定量 控制,在固体药物制剂中的原料药应保持优势药物晶型物质状态,以保证晶型药物产品质量 和临床作用的一致性。由于固体制剂是由复杂成分体系组成,制剂中含各种辅料成分或制剂 工艺可能使原料药晶型发生转变,故需要对固体、半固体、悬浮剂制剂中原料药晶型进行质 量控制,以保证固体制剂中原料药晶型与优势药物晶型一致。 1 药物多晶型的基本概念 用于描述固体化学药物物质状态,由一组参量(晶胞参数、分子对称性、分析排列规律、 分子作用力、分子构象、结晶水或结晶溶剂等)组成。当其中一种或几种参量发生变化而使 其存在有两种或两种以上的不同固体物质状态时,称为多晶型现象(Polymorphism)或称同 质异晶现象。通常,难溶性药物易存在多晶型现象。 固体物质是由分子堆积而成。由于分子堆积方式不同,在固体物质中包含有晶态物质状 态(又称晶体)和非晶态物质状态(又称无定型态、玻璃体) 。晶态物质中分子间堆积呈有序 性、对称性与周期性;非晶态物质中分子间堆积呈无序性。晶型物质范畴涵盖了固体物质中 的晶态物质状态(分子有序)和无定型态物质状态(分子无序) 。 优势药物晶型物质状态可以是一种或多种,故可选择一种晶型作为药用晶型物质,亦可 按一定比例选择两种或多种晶型物质的混合状态作为药用晶型物质使用。 2 晶型样品的制备 采用化学或物理方法,通过改变结晶条件参数可获得不同的固体晶型样品。常用化学方 法主要包括:重结晶法、快速溶剂去除法、沉淀法、种晶法等;常用物理方法主要包括:熔 融结晶法、晶格物理破坏法、物理转晶法等。晶型样品制备方法可以采用直接方法或间接方 法。各种方法影响晶型物质形成的重要技术参数包括:溶剂(类型、组成、配比等) 、浓度、
药品质量控制培训课件
药品质量控制培训教材 培训教师:高行云 2014年1月
第一章总则 1. 为什么要加强质量管理与质量控制的培训:为加强质量检验的规范管理,保证药品质量,贯彻《药品管理法》和《药品生产质量管理规范》等有关法律法规。 2. 质量管理与质量检验贯穿于质量检验部门及药品检验全过程。 3.质保部负责本企业质量检验规范指导工作。 第二章机构、人员与职责 1. 公司应设立质量检验部门(中心化验室)。质量检验部门应配备一定数量的与所生产药品的规模、品种和检验工作相适应的具有专业知识的检验人员。人员职责应明确。 质量检验部门的负责人应具有药学或相关专业大专以上学历,有技术职称;能有效的领导质量检验机构工作,有一定的药品检验和质量管理经验,有能力对药品检验过程中出现的实际问题做出正确的判断和处理;对药品检验质量负全面责任。 从事操作红外分光光度计、气相色谱仪、高效液相色谱仪、薄层色谱扫描仪等大型精密仪器进行原料、辅料、包装材料、半成品及成品检验的人员应具有中专以上药学专业或化学专业学历,经过省或地市级药检所培训,有省食品药品监督管理局颁发培训合格证, 持证上岗; 其中从事滴定液标定的检验人员应具有大专以上药学专业或化学专业学历,或具有中专以上药学专业学历并从事本岗位工作多年,经考核合格, 持证上岗; 从事药品质量检验的其他人员应具有高中以上文化程度,经过专业技术和GMP培训,具有专业基础知识和实际操作技能,具有本岗位检验工作的能力,经岗位考核合格并持证上岗; 从事高生物活性、高毒性、强污染性、高致敏性及有特殊要求的药品质量检验人员应经相应专业的技术培训,具有相关的专业基础知识,并持证上岗; 从事中药材、中药饮片质量验收人员需具有相关的专业知识和识别药材真伪、质量优劣的技能,并持证上岗; 从事实验动物管理和饲养人员应具有初中以上文化程度并接受过专业培训,并持证上岗。 2. 中心化验室的主要职责: 2.1、执行物料、中间产品、成品的法定标准和内控标准,按照药品检验标准操作规程操作,执行留样制度; 2.2、执行检验用设备、仪器、试剂、试液、标准物质、滴定液、检定菌、
如何控制好药品质量及药品生产过程中质量控制点
如何控制好药品质量及药品生产过程中质量控制点药品质量的好坏,直接关系到患者的身体健康和生命安全。因此,药品作为特殊商品对自身质量就有特殊要求,概括说药品质量既要安全有效,又要均一稳定。人们生活水平不断提高,对药品安全的诉求已上升至健康权的高度,国家食品药品监督管理局针对药品生产的质量管理制定了管理规范。然而,近年来不断发生的药品安全事故以及虚假药品等问题,特别是自09年以来相继发生的齐二药假药案件、欣弗不良事件、明胶事件等,使人民群众对用药安全的需求药品产业发展之间的矛盾日益凸现,也使医药行业的社会关注度空前提高,药监部门和药品生产企业面临空前压力。要保障人民群众的用药安全,首先要保证药品质量,众所周知,药品产品质量是药企生产活动的成果,药品质量形成于生产过程。要控制好药品质量,保证药品质量,药品生产企业就必须加强自身的管理。 一.应增强质量意识和责任意识 要控制药品质量,保证药品质量,首先应增强质量意识,因为质量是企业的生命,是企业文化的载体,是企业生存发展的基石。控制药品质量从而保证药品质量既是药品生产企业生存发展的自觉要求,也是药品生产质量管理规范、药品管理法、产品质量法等法律法规对药企的强制要求。因此,作为药品生产企业,作为药品生产企业,要想生存和发展,就必须狠抓质量,没有任何捷径可言。同时,为了更好地控制好药品质量,保证药品质量,药品生产企业还应建立各级人员的质量责任制,明确各级人员的质量责任,定期召开质量分析会,对将要出现的质量风险和已出现的质量问题要及时总结,发现问题、分析问题、解决问题,制定针对性和拓展性解决方案,在实施后观察效果,在没有找出问题的根本原因之前,不得进行生产,确保生产出来的药品无任何潜在的质量隐患。即使在生产过程中没有出现质量问题,也要及时总结,检查既定生产工艺是否需要改进和提高,对现有的各项管理工作进行充实和完善,达到尽善控制好药品质量的目的。 二.加强对生产过程的监督和控制 作为药品生产企业,应当充分理解药品质量是设计、生产出来的,而不是检验出来的深刻内涵。质量管理活动必须贯穿于整个生产过程,以得到符合既定质量标准的产品。药品生产是复杂的过程,从原辅料的进厂到药品生产出来并检验合格放行,涉及到许多工序和环节,在每一个环节都必须把好质量关,把质量不合格的因素和引起质量异常的
化学药物质量控制分析方法验证技术指导原则
化学药物质量控制分析方法验证技术指导原则【】HGPH 5-1指导原则编号: 化学药物质量控制分析方法验证 技术指导原则 二??四年十一月 目录 一、概 述 ..................................................................... ............................................ 1 二、方法验证的一般原 则 ..................................................................... ................ 2 三、方法验证涉及到的三个主要方 面 (2) ,一,需要验证的检测项 目 ..................................................................... . (2) ,二,分析方 法 ..................................................................... .. (3) ,三,验证内 容 ..................................................................... .......................... 3 四、方法验证的具体内 容 ..................................................................... . (3)
,一,专属 性 ..................................................................... (3) 1、鉴别反 应 ..................................................................... (3) 2、杂质检 查 ..................................................................... (4) 3、含量测 定 ..................................................................... (4) ,二,线 性 ..................................................................... . (5) ,三,范 围 ..................................................................... . (5) 1、含量测 定 ..................................................................... (5) 2、制剂含量均匀度...................................................................... (5)
药品生产质量管理规范 GMP 版
药品生产质量管理规范
《药品生产质量管理规范》(Good Manufacture Practice,GMP)是药品生产和质量 管理的基本准则,适用于药品制剂生产的全过程和原料药生产中影响成品质量的。大力推 行药品GMP,是为了最大限度地避免药品生产过程中的污染和交叉污染,降低各种差错的发生,是提高药品质量的重要措施。 ,60年代中开始组织制订药品GMP,则从80年代开始推行。1988年颁布了中国的 药品GMP,并于1992年作了第一次修订。十几年来,中国推行药品GMP取得了一定的 成绩,一批制药企业(车间)相继通过了药品和达标,促进了生产和质量水平的提高。但从 总体看,推行药品GMP的力度还不够,药品GMP的部分内容也急需做相应修改。 国家药品监督管理局自1998年8月19日成立以来,十分重视药品GMP的修订工作,先后召开多次座谈会,听取各方面的意见,特别是药品GMP的实施主体-药品生产企业的 意见,组织有关专家开展修订工作。目前,《药品生产质量管理规范》(1998年修订)已由 国家药品监督管理局第9号局长令发布,并于1999年8月1日起施行。历经5年修订、 两次公开征求意见的《药品生产质量管理规范(2010年修订)》(以下简称新版药品GMP)于2011年3月1日起施行。 内容包括: 目录 第一章总则 1 第二章质量管理 1 第一节原则 1 第二节质量保证 1 第三节质量控制 2 第四节质量风险管理 2 第三章机构与人员 2 第一节原则 2 第二节关键人员 3 第三节培训 4 第四节人员卫生 4 第四章厂房与设施 5 第一节原则 5 第二节生产区 5 第三节仓储区 6 第五章设备 7 第一节原则 7 第二节设计和安装 7 第三节维护和维修 7 第四节使用和清洁 7 第五节校准 8 第六章物料与产品 9
药品生产过程质量风险评估报告模板.doc
药品生产过程质量风险评估报告模板 XXXX胶囊生产过程质量风险管理报告 起草人:审核人:审核人:批准人:质量风险管理号:QRM- 起草日期:审核日期:审核日期:批准日期: XXXX药业有限公司 年月年月年月年月 日日日日 目录 XXXX胶囊生产过程质量风险管理报告 1、简介 2、目的 3、范围 4、引用资料 5、风险管理小组组员及职责分工 6、质量风险管理流程 7、风险管理过程 8、风险管理总结及结论 9、风险管理回顾审核 XXXX胶囊生产过程质量风险管理报告 1.简介: 1.1产品概述:XXXX胶囊为以化学原料药XXXX和适量等辅料制成的化学药胶囊剂制剂,为耳鼻喉科及皮肤科用药类非处方药药品。用于缓解过敏性鼻炎有关的症状,如喷嚏、流涕、鼻痒、鼻塞以及眼部痒及灼烧感。口服药物后,鼻和眼部症状及体征得以迅速缓解。亦适用于缓解慢性荨麻疹、瘙痒性皮肤病及其他过敏性皮肤病的症状和体征。规格为10 毫克;贮藏:遮光,密闭保存;包装:铝塑泡罩包装。每板6粒,每盒1板;每板6粒,每盒2板;每板12粒,每盒1板,每板4粒,每盒1板。有效期:30个月。 1.2生产批量:35万粒,140万粒。 1.3主要生产工艺过程及参数: ,混合速度900转/分,混合时间20分钟;批量为140万粒的预混合和35万粒相同,但原辅料均分4等分进行4次混合。 1.3.3 粘合剂15%聚维酮K30的乙醇溶液的配制:按聚维酮K30:95%乙醇量=1.5:8.5(重
量比)进行配置,溶解完全100目滤布过滤。 ,用GHL-250型高效混合制粒机中(每次35万粒量),设定混合I和切割I开动设备混合10分钟后,徐徐加入粘合剂(15%聚维酮K30的乙醇溶液),加完后继续混合3-4分钟,收集软材。 ,筛网目数为24目。 ,每次干燥量为35万粒胶囊的颗粒量,控制干燥温度为50℃~70℃,干燥时间20分钟。,筛网目数为24目。 ,混合速度900转/分,混合时间20分钟;批量为140万粒的总混:用EYH-1000型二维运动混合机进行,混合时间35分钟(此设备无速度调节)。 ,二号胶囊充填模具,用二号空心胶囊进行充填,装量差异控制在充填量的±8%范围内,充填速度为800~1200粒/分。充填过程中,每20分钟检查一次装量。将充填好的胶囊用HTP-Ⅱ型胶囊、片剂抛光机抛光。 ,同时产品冲印批号、有效期。按4粒/板、6粒/板或12粒/板泡罩规格采用相应的二号胶囊泡罩模具,上、下封温度约为120℃,热封温度为160℃。 ,每个盒子及纸箱均打印产品批号、生产日期、有效期,电子监管码?;按6粒/板×1板/盒×200盒/箱或12粒/板×1板/盒×200盒/箱或6粒/板×2板/盒×200盒/箱或4粒/板×1板/盒×200盒/箱的包装规格进行包装。 1.4中间产品的贮存要求:XXXX胶囊颗粒(总混后)用洁净塑料袋盛装,扎紧袋口置洁净的不锈钢桶中密闭,在口服制剂车间中间站贮存,贮存期:天;XXXX胶囊(批,产品均符合标准规定,未产生过质量事故,也发生过严重的毒副作用及不良反应。 2、目的: 2.1通过对XXXX胶囊生产过程质量风险管理,对其带来的质量风险进行有效控制,保证产品质量。 面进行风险梳理识别。 (1)风险严重程度、发生几率、发现的可能性评分标准及风险级别评判标准及风险控制措施:见下表1. 表2. 表3.表4.
新版药品生产质量管理规范(GMP)最终稿
药品生产质量管理规范国家食品药品监督管理局
电子版目录 第一章总则 (1) 第二章质量管理 (1) 第一节原则 (1) 第二节质量保证 (1) 第三节质量控制 (2) 第四节质量风险管理 (3) 第三章机构与人员 (3) 第一节原则 (3) 第二节关键人员 (3) 第三节培训 (5) 第四节人员卫生 (6) 第四章厂房与设施 (6) 第一节原则 (6) 第二节生产区 (7) 第三节仓储区 (8) 第四节质量控制区 (8) 第五节辅助区 (9) 第五章设备 (9) 第一节原则 (9) 第二节设计和安装 (9) 第三节维护和维修 (10) 第四节使用、清洁和状态标识 (10) 第五节校准 (10) 第六节制药用水 (11) 第六章物料与产品 (11) 第一节原则 (11) 第二节原辅料 (12) 第三节中间产品和待包装产品 (13) 第四节包装材料 (13) 第五节成品 (14) 第六节特殊管理的物料和产品 (14) 第七节其它 (14) 第七章确认与验证 (15) 第八章文件管理 (16) 第一节原则 (16) 第二节质量标准 (17) 第三节工艺规程 (18) 第四节批生产记录 (19) 第五节批包装记录 (20) 第六节操作规程和记录 (20) 第九章生产管理 (21) 第一节原则 (21)
第二节防止生产过程中的污染和交叉污染 (22) 第三节生产操作 (22) 第四节包装操作 (23) 第十章质量控制与质量保证 (24) 第一节质量控制实验室管理 (24) 第二节物料和产品放行 (27) 第三节持续稳定性考察 (28) 第四节变更控制 (29) 第五节偏差处理 (30) 第六节纠正和预防措施 (30) 第七节供应商的审计和批准 (31) 第八节产品质量回顾分析 (31) 第九节投诉 (32) 第十一章委托生产与委托检验 (33) 第一节原则 (33) 第二节委托方 (33) 第三节受托方 (33) 第四节合同 (34) 第十二章产品发运与召回 (34) 第一节原则 (34) 第二节发运 (34) 第三节召回 (34) 第十三章自检 (35) 第一节原则 (35) 第二节自检 (35) 第十四章术语 (35)
药物质量控制基本知识
1.(原始的)色谱图为什么要经过数字化才能成为色谱指纹图谱?色谱指纹图谱数字化包括哪些内容? 一、基本概念 1、指纹图谱(Fingerprint) 指纹图谱是指某种或某产地样品中所共有的、具有特异性的某类或数类成分的色谱或光谱的图谱。其特点在于通过指纹图谱的特异性,能有效鉴别样品的真伪或产地。通过指纹图谱主要特征峰的含量或比例的制定,能有效控制样品的质量,确保样品的相对稳定。 2、数字化色谱指纹图谱(numerical chromatographic fingerprint) 数字化色谱指纹图谱(numerical chromatographic fingerprint)又称相对保留值指纹图谱,其原理是在所有参与比较鉴定样品的色谱图中确定一个在各鉴定样品中都有的色谱峰作为参比标准,然后求取所有色谱峰各自的相对保留值。将色谱峰保留值转化为漂移较少、相对稳定的相对保留值,以此作为色谱峰的定位依据,加上各自的峰面积等参数,构成色谱指纹图谱。然后以这些由色谱图转换成相对保留值的数字化色谱指纹图谱作为标准,对那些参与比较的样品进行研究。二、指纹图谱的发展、属性和作用 指纹图谱的产生与发展[1],指纹图谱技术应用于植物药质盆控制可以追溯到上世纪年代初,随着色谱技术尤其是薄层技术的发展,人们大量采用薄层色谱的方法将植物药中化学成分展开于由各种担体铺成的薄层板上。根据薄层板上斑点的位值、斑点大小、斑点颜色、斑点数目的多少来进行定性鉴别,这种薄层鉴别方法己具备了指纹图谱特征,并被广泛应用。80年代及90年代,由于薄层扫描仪的出现使得薄层鉴别方法得到进一步发展,日本和我国部分学者用当时的薄层扫描仪得到复方成药扫描图谱作为色谱指纹图尝试应用于药材及成药分析。现代新型的薄层扫配备有自动点样系统、成像系统、数据处理系统,可将薄层板色谱结果转变成类于液相的色谱图,薄层扫描色谱图与薄层色谱结果结合,相得益彰,使得薄层扫描术成为指纹图谱研究的主要方法之一。与此同时,高效液相技术得到了长足发展,随着植物药中化学成分的分离鉴定、活性成分的不断阐明,人们更多应用高效液相法行定性、定量分析。西方国家如德国、美国、加拿大等普遍采用高效液相色谱法对物药中己知及未知组分进行控制,并形成相应的规范。新近几年国内有关学术期刊刊登了一些应用高效液相色谱法研究指纹图谱的论文。 何谓中药指纹图谱,谢培山教授将其定义为它是一种综合的,可量化的鉴别
药品生产质量管理规范解析
第二十三条质量管理负责人 (一)资质: 质量管理负责人应当至少具有药学或相关专业本科学历(或中级专业技术职称或执业药师资格),具有至少五年从事药品生产和质量管理的实践经验,其中至少一年的药品质量管理经验,接受过与所生产产品相关的专业知识培训。 (二)主要职责: 1.确保原辅料、包装材料、中间产品、待包装产品和成品符合经注册批准的要求和质量标准; 2.确保在产品放行前完成对批记录的审核; 3.确保完成所有必要的检验; 4.批准质量标准、取样方法、检验方法和其他质量管理的操作规程; 5.审核和批准所有与质量有关的变更; 6.确保所有重大偏差和检验结果超标已经过调查并得到及时处理; 7.批准并监督委托检验; 8.监督厂房和设备的维护,以保持其良好的运行状态; 9.确保完成各种必要的确认或验证工作,审核和批准确认或验证方案和报告; 10.确保完成自检; 11.评估和批准物料供应商; 12.确保所有与产品质量有关的投诉已经过调查,并得到及时、正确的处理; 13.确保完成产品的持续稳定性考察计划,提供稳定性考察的数据; 14.确保完成产品质量回顾分析;
15.确保质量控制和质量保证人员都已经过必要的上岗前培训和继续培训,并根据实际需要调整培训内容。 第二十四条生产管理负责人和质量管理负责人通常有下列共同的职责: (一)审核和批准产品的工艺规程、操作规程等文件; (二)监督厂区卫生状况; (三)确保关键设备经过确认; (四)确保完成生产工艺验证; (五)确保企业所有相关人员都已经过必要的上岗前培训和继续培训,并根据实际需要调整培训内容; (六)批准并监督委托生产; (七)确定和监控物料和产品的贮存条件; (八)保存记录; (九)监督本规范执行状况; (十)监控影响产品质量的因素。 第二十五条质量受权人 (一)资质: 质量受权人应当至少具有药学或相关专业本科学历(或中级专业技术职称或执业药师资格),具有至少五年从事药品生产和质量管理的实践经验,从事过药品生产过程控制和质量检验工作。 质量受权人应当具有必要的专业理论知识,并经过与产品放行有关的培训,方能独立履行其职责。 (二)主要职责: 1.参与企业质量体系建立、内部自检、外部质量审计、验证以及药品不良反应报告、产品召回等质量管
药品晶型研究及晶型质量控制指导原则
9015药品晶型研究及晶型药物的质量控制技术与方法指导原则固体药物及其制剂中存在多晶型现象时,应使用 优势药物晶型物质状态 作为药物原料及其制剂晶型,以保证药品临床有效性二安全性与质量可控性三 当固体药品存在多晶型现象且不同晶型物质状态可影响药品的有效性二安全性与药品质量时,应对固体制剂二半固体二悬浮剂等制剂种类中的原料药晶型物质状态进行定性二定量控制,在固体药物制剂中的原料药应保持优势药物晶型物质状态,以保证晶型药物产品质量和临床作用的一致性三由于固体制剂是由复杂成分体系组成,制剂中含各种辅料成分或制剂工艺可能使原料药晶型发生转变,故需要对固体二半固体二悬浮剂制剂中原料药晶型进行质量控制,以保证固体制剂中原料药晶型与优势药物晶型一致三 当固体药品存在多晶型现象,且不同晶型状态对药品的有效性二安全性或质量可产生影响时,应对药品固体制剂二半固体制剂二混悬剂等中的药用晶型物质状态进行定性或定量控制三药品的药用晶型应选择优势晶型,并保持制剂中晶型状态为优势晶型,以保证药品的有效性二安全性与质量可控三 优势晶型系指当药物存在有多种晶型状态时,晶型物质状态的临床疗效佳二安全二稳定性高等,且适合药品开发的晶型三 1.药物多晶型的基本概念 用于描述固体化学药物物质状态,由一组参量(晶胞参数二分子对称性二分析排列规律二分子作用力二分子构象二结晶水或结晶溶剂等)组成三当其中一种或几种参量发生变化而使其存在有两种或两种以上的不同固体物质状态时,称为多晶型现象(p o l y m o r p h i s m)或称同质异晶现象三通常, 难溶性药物易存在多晶型现象三 固体物质是由分子堆积而成三由于分子堆积方式不同,在固体物质中包含有晶态物质状态(又称晶体)和非晶态物质状态(又称无定型态二玻璃体)三晶态物质中分子间堆积呈有序性二对称性与周期性;非晶态物质中分子间堆积呈无序性三晶型物质范畴涵盖了固体物质中的晶态物质状态(分子有序)和无定型态物质状态(分子无序)三 优势药物晶型物质状态可以是一种或多种,故可选择一种晶型作为药用晶型物质,亦可按一定比例选择两种或多种晶型物质的混合状态作为药用晶型物质使用三 2.晶型样品的制备 采用化学或物理方法,通过改变结晶条件参数可获得不同的固体晶型样品三常用化学方法主要包括:重结晶法二快速溶剂去除法二沉淀法二种晶法等;常用物理方法主要包括:熔融结晶法二晶格物理破坏法二物理转晶法等三晶型样品制备方法可以采用直接方法或间接方法三各种方法影响晶型物质形成的重要技术参数包括:溶剂(类型二组成二配比等)二浓度二成核速率二生长速率二温度二湿度二光度二压力二粒度等三鉴于每种药物的化学结构不同,故形成各种晶型物质状态的技术参数条件亦不同,需要根据样品自身性质合理选择晶型样品的制备方法和条件三 3.晶型物质状态的稳定性 自然界中的固体物质可处于稳定态二亚稳定态二不稳定态三种状态,晶型物质亦如此三化合物晶型物质状态会随着环境条件变化(如:温度二湿度二光照二压力等)而从某种晶型物质状态转变为另外一种晶型物质状态,称为转晶现象三 由于药用晶型物质的稳定性会影响到药品的临床有效性与安全性,故需要对多晶型药物制剂进行晶型物质状态的稳定性研究三研究内容包括:原料药成分的晶型物质状态的稳定性,原料药晶型物质与制剂处方中各种辅料的相容性,制剂的制粒二成型二干燥等工艺对原料药晶型物质状态的影响等三 通过晶型物质状态的稳定性研究,可为优势药物晶型物质状态选择二药物制剂处方二制备工艺过程控制二药品贮存条件等提供科学依据三稳定或亚稳定(有条件的稳定)的晶型物质具有成药性,不稳定晶型物质不具有成药性三根据稳定性试验项下的影响因素试验方法和条件,考察晶型物质状态对高温二高湿二光照条件的稳定性;采用压力方法考察晶型物质状态对压力的稳定性,观察晶型物质状态是否发生转晶现象三 4.晶型药物的生物学评价 需要采用符合晶型物质状态规律的生物学评价的科学方法,溶液状态下的体外细胞评价方法二已发生转晶的悬浮液体内给药等评价方法无法反映固体晶型物质真实的生物学特征三故应采用动物体内试验并固体给药方式,可获得晶型物质真实的生物学评价数据三 5.晶型药物的溶解性或溶出度评价 本法为体外晶型物质评价方法三 当原料晶型物质状态不同时,晶型原料或固体制剂的溶解或溶出性质可能存在较大差异,所以需要进行晶型物质与溶解或溶出性质的关系研究三以溶解度或溶出度二溶解速率或溶出速率作为评价指标三 原料药采用溶解曲线法,固体制剂采用溶出曲线法,可参照‘口服固体制剂溶出度试验技术指导原则“相关内容进行溶解曲线或溶出曲线比较三 6.药品晶型质量研究方法 不同药物的不同晶型物质状态对定性鉴别方法或成分含量定量分析方法的特异性可以相同或不同,方法包含绝对方法和相对方法,可选择有效的质量控制方法三 (1)晶型种类鉴别 定性方法 绝对鉴别方法:可独立完成晶型物质状态鉴别的方法三方法仅适用于晶型原料药三 单晶X射线衍射法(S X R D):属绝对晶型鉴别方法, 四833四 9015药品晶型研究及晶型药物的质量控制技术与方法指导原则
浅谈制药企业生产质量控制
龙源期刊网 https://www.360docs.net/doc/4a3055406.html, 浅谈制药企业生产质量控制 作者:钱慧徐欢 来源:《中国化工贸易·下旬刊》2017年第09期 摘要:本文阐述了生产全过程的质量控制,并明确了药品生产质量控制的重要性,就药 品质量问题影响因素分析,它包括多方面因素,从原辅料采购,到生产过程的控制,再到成品检验合格出厂后,每到工序都需要过程质量控制。因此只有具备严格的管理规范和高技术水平的生产才能能生产出高质量的药品。患者的生命安全才能得到保障。 关键词:质量控制;药品企业;药品质量 药品质量是现在社会关注的重点,因为关系到用药患者的生命安全,目前只有建立完善的质量控制体系,从原料的采购,到生产全过程,到成品出厂,每一个环节,都需要严格的过程控制。也就是说,生产过程质量控制是为了通过监视质量形成过程,消除整个生产环节上所有导致药品安全隐患。以达到降低风险,生产出安全有效的产品。在企业领域,质量控制活动主要是企业内部的生产现场管理,是指为达到和保持质量而进行控制的管理措施和技术措施方面的活动。对生产过程的全程质量控制,主要从以下几方面控制: 1 起点控制 1.1 原辅料控制 对原辅物料进行控制,如购买的原辅料检验不合格,杂质或者含量不够直接影响到最终的产品质量。首先我们物料供应部门应先对原辅料厂家进行供应商审计,如审计合格后,购买原辅料,到货后放入库房待检区,质量监督部门进行取样、检验,入厂后检验合格方可进行生产使用[1]。 1.2 装备设施控制 制药行业与其他的行业不同,由于药品直接危及到用药人群的生命安全,在生产过程中几乎所以的操作都必须由非常专业的药品制造设备进行生产和加工,所以这些设备在生产过程中能够按照设计的要求完成相应的生产任务往往也就影响和控制了药品生产的质量,同时,由于药品生产设备也是容易出现故障的,如果设备比较差,容易影响药品生产,或者物料损失,甚至药品的稳定性,杂质微生物等引入。因此为了保证生产药品的质量可靠性,在这些影响药品质量的众多因素中,制药设备对药品质量的影响是非常突出和至关重要的[2]。 1.3 生产文件、生产人员等控制 生产文件应按GMP要求进行编写,并对生产操作人员进行产期培训。在进入生产现场前按GMP的相关要求进行控制,对不符合规定的应及时纠正或不允许进入生产现场。
USP-化学药物质量控制分析方法验证技术指导原则
1225VALIDATION OF COMPENDIAL PROCEDURES Test procedures for assessment of the quality levels of pharmaceutical articles are subject to various requirements. According to Section 501 of the Federal Food, Drug, and Cosmetic Act, assays and specifications in monographs of the United States Pharmacopeia and the National Formulary constitute legal standards. The Current Good Manufacturing Practice regulations [21 CFR 211.194(a)] require that test methods, which are used for assessing compliance of pharmaceutical articles with established specifications, must meet proper standards of accuracy and reliability. Also, according to these regulations [21 CFR 211.194(a)(2)], users of analytical methods described in USP–NF are not required to validate the accuracy and reliability of these methods, but merely verify their suitability under actual conditions of use. Recognizing the legal status of USP and NF standards, it is essential, therefore, that proposals for adoption of new or revised compendial analytical procedures be supported by sufficient laboratory data to document their validity. The text of this information chapter harmonizes, to the extent possible, with the Tripartite International Conference on Harmonization (ICH) documents Validation of Analytical Procedures and the Methodology extension text, which are concerned with analytical procedures included as part of registration applications submitted within the EC, Japan, and the USA. SUBMISSIONS TO THE COMPENDIA Submissions to the compendia for new or revised analytical procedures should contain sufficient information to enable members of the USP Council of Experts and its Expert Committees to evaluate the relative merit of proposed procedures. In most cases, evaluations involve assessment of the clarity and completeness of the description of the analytical procedures, determination of the need for the procedures, and documentation that they have been appropriately validated. Information may vary depending upon the type of
药品生产质量管理复习题及参考答案
中南大学网络教育课程考试复习题及参考答案 药品生产质量管理 一、填空题: 1.药品GMP的中文全称是,英文全称是,适用范围是。 2.GMP规定批生产记录应按归档,保存至药品有效期后,未规定有效期的药品,其批生产记录至少保 存。 3.从专业化管理角度,GMP可以分为A和B系统,其中A是对 的控制,B是对的保证。 4.实施GMP涉及的三个方面是、和人员,其核心是。 5.我国GMP规定,洁净室布局时要求洁净室等级的静压差为,洁净室与室外大气的静压差为。洁净室无 特殊要求时,温度应控制在度,相对湿度控制在。 6.物料和产品应当根据其性质有序分批贮存和周转,发放及发运应当符合 和的原则;不合格的物料、中间产品、待包装产品和成品的每个包装容器上均应当有清晰醒目的,并在内妥善保存。 7.记录应当保持清洁,和。记录填写的任何更改都应当签注和并使原有信息仍清晰可辨,必要时,应当 说明更改的理由。 8.批记录应当由负责管理,至少保存至药品。质量标准、工艺规程、操作规程、稳定性考察、确认、验 证、变更等其他重要文件应当。 9.应当建立划分产品的操作规程,生产批次的划分应当能够确保同一批次产品质量和特性的。每批产品 应当检查和,确保物料平衡符合设定的限度。 10.药品生产管理部门和质量管理部门的负责人不得_____________。他们应有能力对药品生产和质量管 理中的实际问题作出 ________的________和_______。 11.从事高________活性、高_____性、高________性和强_________性及有特殊要求的生产人员和质检人 员应经相应的技术培训。 12.药品生产企业的______区、______区、______区和_______区的总体布局应合理,不得互相妨碍。 13.洁净区的内表面应平整光滑、接口严密、无_______、无________物脱落,并能耐受________和 ________。 14.设备的设计、选型、安装、改造和维护必须符合用途,应当尽可能降低___________、___________、 ___________和___________的风险,便于操作、清洁、维护,以及必要时进行的______或______。15.设备与药品直接接触的表面应当平整、光洁、易清洗或消毒、耐腐蚀,不得 _____、 ___________或___________。 16.不得使用__________、__________、___________的衡器、量具、仪表以及用于记录和控制的设备、 仪器。 17.模具使用后,由________归还至模具间,由_______或_______检查模具完好及清洁情况。 18.质量管理负责人和生产管理负责人不得互相兼任。和可以兼任。应当制定操作规程确保独立履行职 责,不受和其他人员的干扰。 19.企业应当指定部门或专人负责培训管理工作,应当有经或质量管理负责人培训方案或计划,应当予以 保存。 20.与药品生产、质量有关的所有人员都应当经过,培训的内容应当与的要求相适应。除进行本规范理论 和实践的培训外,还应当有相关法规、相应岗位的、的培训,并培训的实际效果。 21.生产设备应有明显的状态标识,标明和(如名称、规格、批号);没有内容物的应当标明。 22.采用新的生产处方或生产工艺前,应当验证其。生产工艺在使用规定的和 条件下,应当能够始终生产出符合预定用途和的产品。 23.应当根据确认或验证的对象制定确认或,并经、。确认或验证方案应当。 24.质量控制实验室的检验人员应当具有相关专业学历,并经过与所从事的检验操作相关的且。 25.试液和已配制的培养基应当标注配制、和配制人员姓名,并有配制(包括灭菌)记录。不稳定的试 剂、试液和培养基应当标注及。标准液、滴定液还应当标注最后一次标化的日期和校正因子,并有标化记录。
药品生产过程质量风险产生的原因及控制
药品生产过程质量风险产生的原因及控制 发表时间:2019-10-29T13:46:00.847Z 来源:《健康世界》2019年13期作者:姬广欣王丽萍徐瑞朋 [导读] 对于食品药品生产企业以及广大消费者都具有十分重要的意义。 山东裕欣药业有限公司山东省临沂市 276000 摘要:随着时代的进步人们对生活质量有了更高要求。食品与药品是我们日常生活中的必需品,因此,这两类产品的性质注定了其生产的企业要有一个高质量的生产流程。本文主要针对食品药品质量风险管理进行了概述,分析了食品药品生产过程中质量风险产生的原因,并根据实际情况,提出了几种食品药品生产过程质量风险的控制方法,希望能够在一定程度上降低食品药品生产过程中的质量风险,对于食品药品生产企业以及广大消费者都具有十分重要的意义。 关键词:食品药品生产;质量风险;原因与控制 引言 药品生产的质量保证需要完善科学的检验方法,也是要对药品生产的整个过程展开严格的控制,药品生产的各个环节保持合理性,对保证药物质量是有直接影响的。在药品生产的实际开展中,存在诸多对药物质量产生影响的因素,因此需要在药品生产中加强对各个环节的监督,并对各类风险的因素展开分析,建立完善和科学的质量控制体系,为药品生产提供科学的依据,让人们的生命财产可以得到更加完善的保障,也是为了让医药行业可以更加健康地发展起来,为人类的进步提供助力。 1药品质量风险管理概述 质量风险管理,是在产品生产的整个周期内始终贯穿的一种机制,在药品生产中,进行整个生产周期的质量评估、控制以及审核。药品生产的质量管理是非常重要的过程,风险分析也是涉及到诸多的因素,评估公司的控制措施是否足够完善,对于中高风险是否有降低的有效措施,对风险发生有没有预防的措施等。这些都是对药品生产的质量控制有直接的影响。而药品生产中的质量风险控制,主要是在药品的生产过程中进行综合性监督管理过程,对疗效以及安全进行管控。风险控制是为了对治疗疾病提供更加合理的保障,同时也是对药品的一种基本需求。在药品生产中,本身是起到防病和治病的作用,药品的质量管控是复杂的,在研发、生产以及经营等各个领域,涉及到的影响因素众多。 2药品生产过程质量风险产生的原因 2.1食品药品生产管理存在问题 食品药品生产管理中主要存在以下几个方面的问题。(1)部分企业负责人缺乏质量风险意识。有一些企业在生产过程中偷工减料,不能持续生产合格的产品。还有一些企业在创业初期能够保证产品的质量,但是随着企业发展,有了一定的基础及知名度,由于产品需求量变大,企业这个时候就容易出现动摇,偏离初衷,这样生产出的产品就很容易不合格。(2)一些企业的生产工艺执行欠缺。要想生产出高质量的产品,标准的工艺操作流程是关键,标准的工艺操作流程可以为食品及药品的生产过程提供重要保障。然而,一些企业并没有严格执行工艺操作流程,甚至存在擅自更改生产流程的现象,从而造成生产过程失控,这样生产出来的产品注定质量不过关。 2.2化学制药工艺存在的问题 化工制药涉及到的问题非常明显,主要涉及化学合成工艺路线或者试剂、溶媒、参数控制是否是目前最优的路线。在化学合成的环节,工艺路线的合理性,各方面参数的控制,很多并未达到相关标准,一些设备无法满足药物检验以及控制,或者是化工制药设备无法满足工艺的要求。而且化工制药本身是涉及到高排放以及高污染的问题。一些药品生产中制药工艺的水平比较低,自动化生产程度不高。对药品生产中的各类参数缺乏控制,很多缺乏科学以及统一的控制标准,导致了药品批次存在诸多的差异性。在药品生产中这些问题都是对现有化工制药的工艺造成一定的挑战,对于化工制药企业来说,找到适合自己的工艺方案,并提升自身的工艺水平是重中之重。 2.3药物自身特殊性 药品生产本身涉及到药物的特殊性,药品生产的原料中,包括各类原材料以及辅料,质量和品种随着供应商不同以及工艺不同,也是有着极大的差别,在药品生产中的包装材料也是因为各企业的标准有所差异,对药品生产质量有所影响。从工艺上分析,药品生产的工艺涉及到各个环节。让药品生产更加复杂,药物自身的特殊性,则是因为现阶段各类药物的种类繁多。在其中某个环节若是出现问题,都是会导致药品质量出现严重的问题。 3质量风险控制措施 3.1建立和完善风险管理体系 在确保产品质量和适宜的工业性能基础上,建立和完善控制系统的生产风险管理系统,推行质量授权人制度,确保每批上市销售的产品均能够符合上市许可法规和GMP规定。明确专业技术人员在生产质量管理过程中的主导地位,将最终责任落实到授权人,促进企业建立明确、清晰的管理框架。通过建立切实可行的生存过程质量风险控制体系,健全企业内控质量标准,晚上物料管理制度,制定详尽的生存过程执行标准化文书,规范产品审核放行管理制度文件,建立健全质量管理系统。同时通过相关培训,提高员工质量风险意识。 3.2采用新的生产技术 上面讲到,食品药品生产过程中质量风险产生的一个原因是生产的技术水平不够。因此,要提高质量风险管理的水平,首先必须要提高食品药品的生产技术水平,尽量采用先进的生产技术;其次可以加强过程分析技术的应用,从生产技术、生产工艺上提高药品的质量。过程分析技术可以实时测量药品生产过程的关键参数,这样一旦发生情况,也容易及时检查与修整;另外,还需要采用先进的过程分析仪器与过程控制工具,对生产过程进行详尽的分析,确定质量控制的关键步骤与关键环节,尽最大可能消除药品质量风险。 3.3发挥药政管理部门的导向作用 现阶段药品生产的风险管理,是国家药品监管的重要方向,发达国家不断完善药品生产监管体系,并成立了相关的管理机构,国内的药品监管一直以来是依靠行政监管,并未对企业在药品生产质量管理方面进行针对性的技术指导。还是需要将行政部门的导向作用充分发挥出来,不断完善企业如何开展药品生产风险管理的制度以及评估指南,结合国内的实际情况,在药品生产的监管方面要建立更加完善和