上海医疗器械销帮主软件使用说明精品篇

上海三类医疗器械销帮主软件操作说明
-----10年工作经验的有详细步骤的教学版本超级实用。
食药监检查需要检查三类医疗器械的3大软件:
第一:不良系统
注册不良检查系统https://www.360docs.net/doc/5c5243698.html,/console/login.ftl 用户名和密码一定要记住,否则追回很麻烦,要写书面报告
需要自己在网站注册。
第二:追溯系统
注册不良检查系统https://www.360docs.net/doc/5c5243698.html,/login
医疗器械的年报就是在这个网站提交,每年的1月份需要提交年报信息。
需要自己在网站注册。
第三:销帮主软件
销帮主软件,https://www.360docs.net/doc/5c5243698.html,/
需要联系办理三类医疗许可证的公司购买,需要随时把进销存的记录录入,每年检查一次。
这个三个软件中最难的就是销帮主软件的使用。
今天详细的说明下销帮主软件使用,没有基础的人看完本文也可以看得懂。
大家需要记住,在销售软件里,所有的录入完成,都要进行查询,然后选中刚才录入的记录,进行审核。只有审核了录入的记录,才能进行下一步,否则下一步是没办法操作,这个是销帮主系统最难的一个地方,需要大家特别注意。
没有审核的时候,在查询的时候,刚才录入的记录前面有铅笔的符号,可以删除,你审核后,就没有铅笔的符号,就不能删除了。
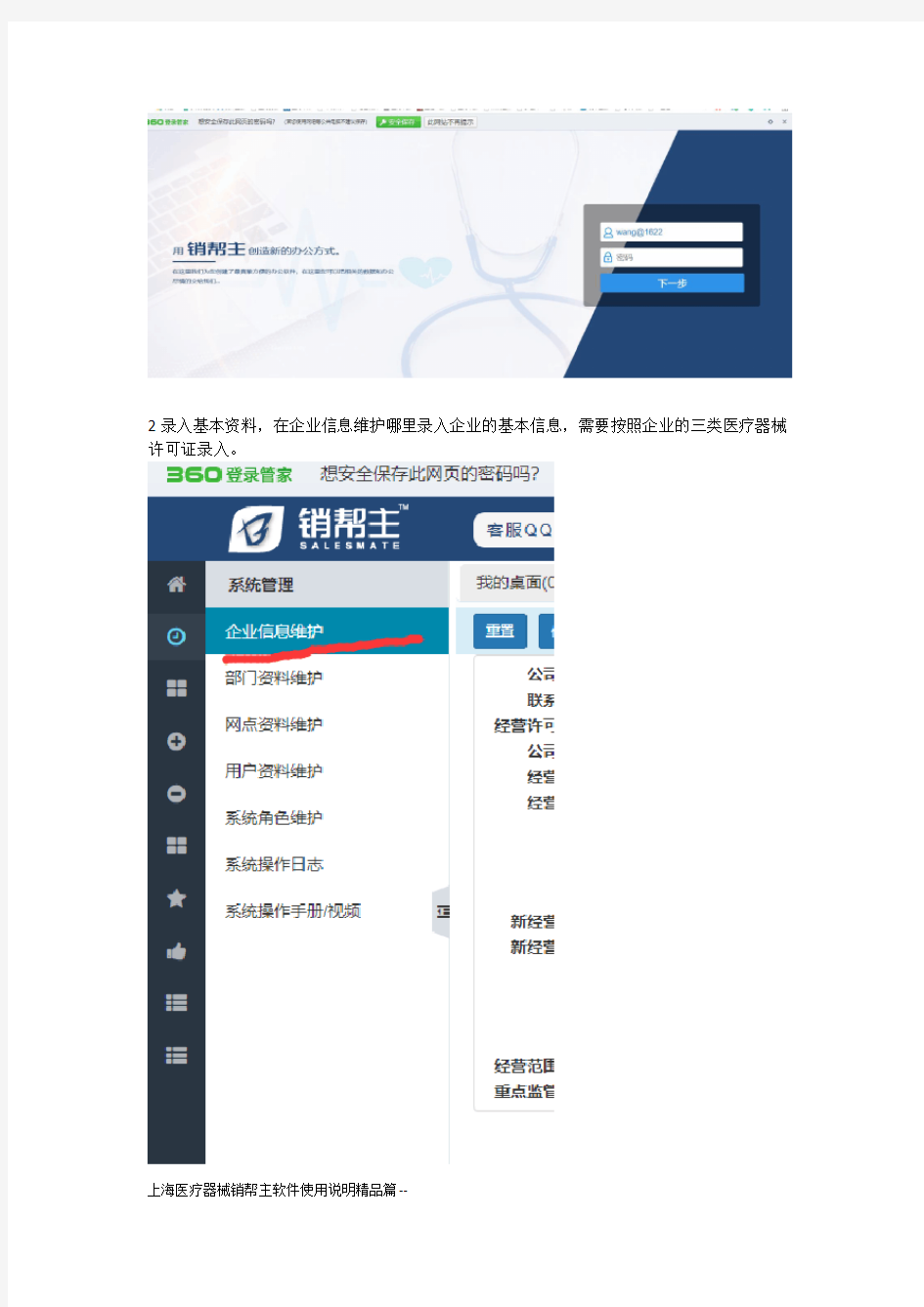
1登陆销帮主,打开网站,输入用户名和密码。
2录入基本资料,在企业信息维护哪里录入企业的基本信息,需要按照企业的三类医疗器械许可证录入。
3开始录入基本资料,基本资料分成3个部分,
新公司销帮主软件的操作流程。
1供货企业的资本资料。
供货企业档案维护-----录入基本资料-需要供应商的执照和供应商的三类医疗许可证
供应商的法人销售授权书。需要在基本资料录入的时候,点击编辑—补充录入。
供货企业质保协议。--需要在供货企业质保协议哪里录入上传附件。
2购货企业的基本资料,
购货企业档案维护—录入基本资料,需要购货企业,资质,如果是公司需要三类许可证。如果是医院客户,需要医院提供医疗机构执业许可证。
需要销售公司负责这个客户的授权书。--需要在基本资料录入的时候,点击编辑—补充录入。
3商品档案管理(开票产品的生产许可证)
商品分类信息—新增分类编码-和分类名称,分类编码0001到9999 分类名称按商品的注册证的拼音的首个字母。
商品的档案维护—就是根据开票的商品-录入商品注册证上的信息。
4首营审核,3次流转,1次审核。
医疗器械注册申报资料要求和批准证明文件格式
中华人民共和国医疗器械注册证 (格式) 注册证编号: (审批部门盖章)
中华人民共和国 医疗器械注册变更文件 (格式) 注册证编号: 批准日期:年月曰 (审批部门盖章)
国家食品药品监督管理总局医疗器械临床试验批件 (格式) 批件号:
日 (审批部门盖章) 附件4 医疗器械注册申报资料要求及说明
注册申报资料应有所提父资料目录,包括申报资料的一级和一级 标题。每项二级标题对应的资料应单独编制页码。 一、申请表 二、证明性文件 (一)境内申请人应当提交: 1. 企业营业执照副本复印件和组织机构代码证复印件。 2. 按照《创新医疗器械特别审批程序审批》的境内医疗器械申请 注册时,应当提交创新医疗器械特别审批申请审查通知单,样品委托其他企业生产的,应当提供受托企业生产许可证和委托协议。生产许
可证生产范围应涵盖申报产品类别 (二)境外申请人应当提交: 1. 境外申请人注册地或生产地址所在国家(地区)医疗器械主管部门出具的允许产品上市销售的证明文件、企业资格证明文件。 2. 境外申请人注册地或者生产地址所在国家(地区)未将该产品作为医疗器械管理的,申请人需要提供相关证明文件,包括注册地或者生产地址所在国家(地区)准许该产品上市销售的证明文件。 3. 境外申请人在中国境内指定代理人的委托书、代理人承诺书及营业执照副本复印件或者机构登记证明复印件。 三、医疗器械安全有效基本要求清单 说明产品符合《医疗器械安全有效基本要求清单》(见附件8)各项适用要求所采用的方法,以及证明其符合性的文件。对于《医疗器械安全有效基本要求清单》中不适用的各项要求,应当说明其理由。 对于包含在产品注册申报资料中的文件,应当说明其在申报资料中的具体位置;对于未包含在产品注册申报资料中的文件,应当注明该证据文件名称及其在质量管理体系文件中的编号备查。 四、综述资料 (一)概述 描述申报产品的管理类别、分类编码及名称的确定依据。
医疗器械注册管理办法4号令
医疗器械注册管理办法 国家食品药品监督管理总局令第4号 国家食品药品监督管理总局令 第 4 号 《医疗器械注册管理办法》已于2014年6月27日经国家食品药品监督管理总局局务会议审议通过,现予公布,自2014年10月1日起施行。 局长张勇 2014年7月30日 医疗器械注册管理办法 第一章总则 第一条为规范医疗器械的注册与备案管理,保证医疗器械的安全、有效,根据《医疗器械监督管理条例》,制定本办法。 第二条在中华人民共和国境内销售、使用的医疗器械,应当按照本办法的规定申请注册或者办理备案。 第三条医疗器械注册是食品药品监督管理部门根据医疗器械注册申请人的申请,依照法定程序,对其拟上市医疗器械的安全性、有效性研究及其结果进行系统评价,以决定是否同意其申请的过程。 医疗器械备案是医疗器械备案人向食品药品监督管理部门提交备案资料,食品药品监督管理部门对提交的备案资料存档备查。 第四条医疗器械注册与备案应当遵循公开、公平、公正的原则。 第五条第一类医疗器械实行备案管理。第二类、第三类医疗器械实行注册管理。 境内第一类医疗器械备案,备案人向设区的市级食品药品监督管理部门提交备案资料。 境内第二类医疗器械由省、自治区、直辖市食品药品监督管理部门审查,批准后发给医疗器械注册证。 境内第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。 进口第一类医疗器械备案,备案人向国家食品药品监督管理总局提交备案资料。 进口第二类、第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。 香港、澳门、台湾地区医疗器械的注册、备案,参照进口医疗器械办理。 第六条医疗器械注册人、备案人以自己名义把产品推向市场,对产品负法律责任。
省医疗器械注册人制度试点实施方案【模板】
XX省医疗器械注册人制度试点实施方案 (征求意见稿) 为推进XX省医疗器械创新、高质量发展,全面提升我省医疗器械产业化发展水平,依据中共中央办公厅国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》、国务院《关于印发6个新设自贸试验区总体方案的通知》和国家药品监督管理局《关于扩大医疗器械注册人制度试点工作的通知》的相关要求,结合我省工作实际,制定本实施方案。 一、总体目标 鼓励医疗器械创新,优化资源配置,促进注册、生产跨区域产业链发展。探索医疗器械审评审批和委托生产管理新模式,探索建立医疗器械监管协同发展机制,构建跨区域协同合作的医疗器械监管格局。落实注册人全生命周期法律责任,完善事中事后监管体系,积累注册人制度试点经验,为全面实施注册人管理制度奠定基础。 二、基本原则 医疗器械注册人制度是指医疗器械注册申请人(以下简称申请人)提出医疗器械上市许可申请,其样品委托受托生产企业生产并获得医疗器械注册证后,成为医疗器械注册人(以下简称注册人);注册人委托受托生产企业生产产品,以注册人名义上市,并对医疗器械设计开发、临
床试验、生产制造、销售配送、售后服务、产品召回、不良事件监测与再评价等全生命周期产品质量承担相应法律责任的制度。 (一)依法依规,有序推进。贯彻中共中央办公厅国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》精神,落实《关于扩大医疗器械注册人制度试点工作的通知》要求,根据《医疗器械监督管理条例》等法规规定,依法依规、有序开展试点工作。 (二)风险可控,权责明确。坚持风险全程可控,注册人、受托生产企业、监管部门权责明确,落实医疗器械全生命周期监管,确保医疗器械的安全有效。 (三)有效衔接,协调合作。强化医疗器械上市许可和事中事后监管的有效衔接,加强与其他试点省份的沟通协调,构建跨省监管协作机制,建立统一协调合作而又分工责任明确的医疗器械注册人监管体系。 (四)改革创新,全力推动。创新思维、先行先试,破解难题,积极推进试点工作。通过总结评估,形成可复制、可推广的试点经验和创新制度。 三、主要内容 (一)住所或者生产地址位于XX省内的企业、科研机构可以成为申请人,申请医疗器械产品注册。申请人可以委托XX省内或其他试点省份中具备相应生产能力的企业生产样品。
医疗器械注册与备案管理办法.doc
医疗器械注册与备案管理办法 (征求意见稿) 第一章总则 第一条为规范医疗器械的注册与备案管理,保证医疗器械的安全、有效,根据《医疗器械监督管理条例》,制定本办法。 第二条在中华人民共和国境内销售、使用的医疗器械应当按照本办法的规定申请注册或者办理备案。 第三条医疗器械注册是指食品药品监督管理部门根据医疗器械注册申请人的申请,依照法定程序,对其拟上市销售医疗器械的安全性、有效性研究及其结果进行的系统评价,以决定是否同意其申请的审批过程。 第四条医疗器械备案是指食品药品监督管理部门对医疗器械备案人提交的第一类医疗器械备案资料存档备查。 第五条医疗器械注册与备案应该遵循公开、公平、公正的原则。 第六条第一类医疗器械实行备案管理。第二类、第三类医疗器械实行注册管理。 境内第一类医疗器械由设区的市级食品药品监督管理部门予以备案。 境内第二类医疗器械由省、自治区、直辖市食品药品监督管理部门审查,批准后发给医疗器械注册证。 境内第三类医疗器械由国家食品药品监督管理总局审查,
批准后发给医疗器械注册证。 进口第一类医疗器械由国家食品药品监督管理总局予以备案。 进口第二类、第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。 香港、澳门、台湾地区医疗器械的注册、备案,参照进口医疗器械办理。 第七条食品药品监督管理部门应当建立信息系统,公示审批过程和审批结果,供公众查询。 第八条国家鼓励医疗器械的研究与创新,促进医疗器械新技术的推广与应用,推动医疗器械产业的发展。创新医疗器械特别审批程序由国家食品药品监督管理总局另行制定。 第二章基本要求 第九条医疗器械注册申请人(以下简称申请人)是指提出医疗器械注册申请,在该申请获得批准后持有注册证,并以自己名义把产品推向市场,对产品负法律责任的企业。 医疗器械备案人(以下简称备案人)是指办理医疗器械备案,并以自己名义把产品推向市场,对产品负法律责任的企业。 申请注册或者办理备案事务的人员应当具有相应的专业知识,熟悉医疗器械注册或者备案管理的法律、法规、规章和技术要求。 第十条申请人或者备案人在申请注册或者办理备案前,应当遵循医疗器械安全有效基本要求,完成医疗器械的研制,
医疗器械注册管理法规解读
医疗器械注册管理法规解读之一(《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》部分) 2015年02月05日 一、《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》修订的总体思路和原则是什么? 依据《条例》设定的原则和要求对《办法》进行修订。修订的总体思路与《条例》修订的总体思路保持一致,以分类管理为基础,以风险高低为依据,确定医疗器械注册与备案的具体要求。医疗器械注册是一项行政许可制度,是食品药品监督管理部门根据医疗器械注册申请人的申请,依照法定程序,对其拟上市医疗器械的安全性、有效性研究及其结果进行系统评价,以决定是否同意其申请的过程。医疗器械备案是医疗器械备案人向食品药品监督管理部门提交备案资料,食品药品监督管理部门对提交的备案资料存档备查。对第一类医疗器械备案管理,是基于产品风险的考虑设置的行政监管手段。备案人向行政机关报送资料,行政机关对备案资料进行形式审查,发给备案人备案凭证,并公布备案信息。通过备案存档收集信息并
开展后续监督检查,对不合乎法规要求的,应责成企业及时纠正或采取行政处罚等行政行为。 二、医疗器械备案和注册需分别向哪些部门申请? 第一类医疗器械实行备案管理。第二类、第三类医疗器械实行注册管理。境内第一类医疗器械备案,备案人向设区的市级食品药品监督管理部门提交备案资料。境内第二类医疗器械由省、自治区、直辖市食品药品监督管理部门审查,批准后发给医疗器械注册证。境内第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。进口第一类医疗器械备案,备案人向国家食品药品监督管理总局提交备案资料。进口第二类、第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。香港、澳门、台湾地区医疗器械的注册、备案,参照进口医疗器械办理。 三、申请医疗器械备案需提交的资料? 第一类医疗器械备案需提交安全风险分析报告、产品技术要求、产品检验报告、临床评价资料、产品说明书及最小销售单元标签设计样稿、生产制造信息、证明性文件、符合性声明等资料。 四、申请医疗器械注册需提交的资料? 申请医疗器械注册需提交申请表、证明性文件、医疗器械
医疗器械上市公司名单
中国医疗器械上市公司 601607 上海医药部分医疗器械上海1994 600055 万东医疗X放射北京1997.5 600587 新华医疗消毒灭菌X放射山东2002.9 600718 东软股份医用影像 600763 通策医疗口腔医疗浙江1996 600196 复兴医药8个医疗器械公司上海 600288 大恒科技放疗系统北京2000 600661 新南洋上海交大南洋医疗器械公司上海1993 600056 中国医药部分医疗器械销售、北京 600079 人福医药药品、计生用品器械武汉1997.6 600690 青岛海尔制冷设备洁净柜山东1993 600216 浙江医药部分医疗器械销售浙江1999.10 600855 航天长峰麻醉机北京2005.1 中小企业板: 002016 世荣兆业威尔科技超声波器械广东2004 002030 达安基因医学检验、病理检验、试剂广东2004.8 002432 九安医疗电子血压计、低频治疗仪生产天津2010
000603*ST 威达广东1996 002223 鱼跃医疗医疗设备上海2008.4 002022 科华生物检验、生化分析仪、试剂上海2004 000566 海南海药主营医药人工耳蜗海南1994 002144 宏达高科威德耳B 超浙江2007.8 000676 思达高科其中两个公司有医疗影像河南1996.12 000423 东阿阿胶医药体温计、血压计山东1996.7 000705 浙江震元医药、两个公司有器械销售浙江1997 创业板 300003 乐普医疗支架北京2009.10 300030 阳普医疗实验室设备广州2009.12 300015 爱尔眼科眼科医院湖南2009.10 00325HK创生控股生产及销售骨科植入器械。江苏2005 00233HK 铭源医疗蛋白芯片体检上海2002 00853HK微创医疗支架、骨科器械上海2010 08143HK华夏医疗综合医院 00648HK 中国仁济医疗伽玛刀等肿瘤设备租赁香港 08116HK 中国公共医疗医院管理软件香港2007 00801HK 金卫医疗采写输血设备耗材、血液科医院香港2001 08130HK 杏林医疗信息国康医疗器材公司
医疗器械注册专员管理制度
医疗器械注册专员管理制度一、医疗器械注册专员是指经医疗器械生产 企业推荐且熟悉医疗器械相关法律法规和规定要求(含医疗器械注册申报程序)的从事医疗器械注册申报工作的相关人员。 二、各医疗器械生产企业可根据业务需要自愿确定医疗器械注册专员的人数,一般情况为1—3名,要求人员相对固定。 三、申报医疗器械注册专员的人员(以下简称申请人)应具备以下条件: (一) 大专以上学历或初级以上技术职称; (二) 具有2年以上(含2年)相关工作经验或专业背景; (三) 从事医疗器械注册工作的在岗人员。 四、申请人应由聘用单位推荐,填写《湖南省医疗器械注册专员登记表》,经聘用单位签署意见、盖章后,报我局医疗器械处。 五、医疗器械注册专员有以下职责和权力: (一)遵守国家有关法律法规、管理规定和职业道德规范; (二)按照国家食品药品监督管理局或湖南省食品药品监督管理局的有关规定准备、审查、补充和完善医疗器械注册申报资料,保证申报资料的完整规范,并按规定程序完成申报手续; (三)关注国家食品药品监督管理局和湖南省食品药品监督管理局网站发布的有关信息,及时掌握医疗器械注册相关政策和有关医疗器械品种及国家、行业标准的最新动态; (四)有权拒绝办理申报单位申报资料不完整、不规范、不真实、手续不齐全的医疗器械注册事项,维护申报单位的合法权益; ; 接受医疗器械法规及业务培训,不断提高医疗器械注册业务水平)五( — 1 — (六)医疗器械注册专员在申报产品注册中不得弄虚作假。 六、聘用单位应加强对医疗器械注册专员的培养和管理,每年对医疗器械注册专员进行必要的业务培训。 七、医疗器械注册专员要接受我局的监督管理和业务指导、培训,逐步完善医疗器械注册专员诚信管理制度。并为医疗器械注册专员提供定期或不定期的培训,并及时通报医疗器械注册相关信息。 八、我局医疗器械处负责医疗器械注册专员的管理工作。 九、本办法自发布之日起试行。 — 2 — 附表: 湖南省医疗器械注册专员登记表 姓名性别 年龄专业 学历职称 联系电话手机
医疗器械注册管理规定
医疗器械注册管理规定Prepared on 21 November 2021
国家食品药品监督管理总局令 第 4 号 《医疗器械注册管理办法》已于2014年6月27日经国家食品药品监督管理总局局务会议审议通过,现予公布,自2014年10月1日起施行。 局长张勇 2014年7月30日 医疗器械注册管理办法 第一章总则 第一条为规范医疗器械的注册与备案管理,保证医疗器械的安全、有效,根据《医疗器械监督管理条例》,制定本办法。 第二条在中华人民共和国境内销售、使用的医疗器械,应当按照本办法的规定申请注册或者办理备案。 第三条医疗器械注册是食品药品监督管理部门根据医疗器械注册申请人的申请,依照法定程序,对其拟上市医疗器械的安全性、有效性研究及其结果进行系统评价,以决定是否同意其申请的过程。医疗器械备案是医疗器械备案人向食品药品监督管理部门提交备案资料,食品药品监督管理部门对提交的备案资料存档备查。 第四条医疗器械注册与备案应当遵循公开、公平、公正的原则。 第五条第一类医疗器械实行备案管理。第二类、第三类医疗器械实行注册管理。 境内第一类医疗器械备案,备案人向设区的市级食品药品监督管理部门提交备案资料。境内第二类医疗器械由省、自治区、直辖市食品药品监督管理部门审查,批准后发给医疗器械注册证。境内第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。进口第一类医疗器械备案,备案人向国家食品药品监督管理总局提交备案资料。进口第二类、第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。香港、澳门、台湾地区医疗器械的注册、备案,参照进口医疗器械办理。 第六条医疗器械注册人、备案人以自己名义把产品推向市场,对产品负法律责任。
医疗器械公司大全
12月5日入围企业名单电生理类耗材 杭州博杰医学科技有限公司 深圳市惠泰医疗器械有限公司 上海帕斯医疗器材贸易有限公司 美敦力医疗用品技术服务(上海)有限公司(起搏器) 圣犹达医疗用品(上海)有限公司 强生(上海)医疗器材有限公司 脊柱创伤类耗材 北京威联德骨科技术有限公司 上海凯利泰医疗科技有限公司 史赛克(北京)医疗器械有限公司 上海睿星基因技术有限公司 广州幸好医疗器械有限公司 通用(上海)医疗器材有限公司 上海安久生物科技有限公司 长沙德迈医疗器械有限公司 天津市新中医疗器械有限公司 上海贝奥路生物材料有限公司 天津中津生物发展有限公司 厦门大博颖精医疗器械有限公司 百赛国际贸易(上海)有限公司 宁波江东诺邦医疗器械有限公司 湖北联结生物材料有限公司 常州市康辉医疗器械有限公司 北京市同日升医疗器械有限公司 广州爱克曼器械设备有限公司 山东冠龙医疗用品有限公司 柯思摩国际贸易(上海)有限公司 常州亨杰医疗器械有限公司 苏州市康力骨科器械有限公司 苏州海欧斯医疗器械有限公司 苏州艾迪尔医疗器械有限公司 深圳市光明创博生物制品发展有限公司 上海中智医疗器械有限公司 北京市津威康达医疗器械有限公司 北京鑫康辰医学科技发展有限公司 天津市威曼生物材料有限公司 上海迈凯医疗器械有限公司 北京市富乐科技开发有限公司 北京飞渡医疗器械有限公司 苏州欣荣博尔特医疗器械有限公司 成都迪康中科生物医学材料有限公司 苏州爱得科技发展有限公司 上海昕昌记忆合金科技有限公司
深圳市生物桥科技开发有限公司 山东威高骨科材料有限公司 四川大学生物材料工程研究中心 法国LDR医疗公司北京代表处 北京理贝尔生物工程研究所 兰州西脉记忆合金股份有限公司 广州市晟泽贸易有限公司 天津市金兴达实业有限公司 上海医疗器械(集团)有限公司手术器械厂常州奥斯迈医疗器械有限公司 武汉德骼拜尔外科植入物有限公司 上海迪索泰医疗器械有限公司 北京百慕航材高科技股份有限公司 北京百优普泰医疗品有限公司 上海格乐医疗器械有限公司 强生(上海)医疗器材有限公司(脊柱创伤) 常州邦德医疗器械有限公司 上海逸通实业有限公司 辛迪思(上海)医疗器械贸易有限公司 北京市奥斯比利克新技术开发有限公司捷迈(上海)医疗国际贸易有限公司 创生医疗器械(江苏)有限公司 珠海港康达医疗器材有限公司 天津正天医疗器械有限公司 浙江广慈医疗器械有限公司 施乐辉医用产品国际贸易(上海)有限公司北京市春立正达科技开发有限公司 上海浦卫医疗器械厂 强生(上海)医疗器材有限公司(关节) 上海典可医疗科技有限公司 人工关节 联贸医疗用品技术(上海)有限公司 北京威联德骨科技术有限公司 史赛克(北京)医疗器械有限公司 美精技医疗器械(上海)有限公司 深圳市博恩医疗器材有限公司 上海复升医疗器械有限公司 北京力达康科技有限公司 上海迈凯医疗器械有限公司 苏州欣荣博尔特医疗器械有限公司 北京爱康宜诚医疗器材有限公司 北京金查理人工关节技术有限公司 北京蒙太因医疗器械有限公司 强生(上海)医疗器材有限公司(关节)
英文版进口医疗器械注册申请人和备案人名称使用中文的公告
CFDA Notice about Using Chinese Description in Name of the Registration Applicant and the Filing Applicant for ImportedMedical Device (2017 No.131) According to Regulations for the Supervision and Administration of Medical Devices, Provisions for Medical Device Registration,Provisions for In-vitro Diagnostic Reagent Registration and Provisions for Instructions and Labels of Medical Devices and so on regulations and provisions, medical devices that are applying for being marketed in China, shall use Chinese description in the registration applicant name.In order to implement the relevant requirements further, meet the needs of the public better, and accept the social supervision, currently, the notice of the relevant matters that using Chinese description in the registration applicant name is shown as below (hereinafter theregistration applicant, the registrantand the filing applicant of the imported medical device are called by a joint name as "enterprise"): I. The using principle of Chinese (1) The Chinese enterprise name shall use simplified Chinese characters. The Chinese shall be translated by the enterprise itself according to the character translation principle. (2) The Chinese enterprise name shall correspond to the original name,its content shall not be added or deleted. The same enterprise shall use the same Chinese enterprise name. (3) Chinese enterprise name shall not contain content and words that are detrimental to the state or the public interest, that are likely to lead the public to be deceived or misunderstood, as well as that are forbidden from other laws, regulations and provisions. Chinese name should not be changed if the original name was unchanged.If the Chinese name must be changed according to law and regulation, the registration administrative matters change or the filing information change shall be conducted. II. Relevant procedures and the submission documents requirements
吉林省医疗器械注册人制度试点工作实施方案(试行)
吉林省医疗器械注册人制度试点工作实施方案(试 行) 为深入贯彻落实中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)、《国务院关于中韩(长春)国际合作示范区总体方案的批复》(国函〔2020〕45号)、国家药品监督管理局《关于扩大医疗器械注册人制度试点工作的通知》(国药监械注〔2 019〕33号)、《国家药监局关于争取中韩(长春)国际合作示范区相关政策支持的复函》(药监综科外函〔2019〕628号)等文件精神,在吉林省实施医疗器械注册人(以下简称注册人)制度试点工作,结合实际,制定本方案。 一、总体目标 通过在全省开展注册人制度试点工作,探索建立注册人委托生产管理制度,优化资源配置,减少重复建设,激发产业创新发展活力,促进高科技医疗器械成果快速转化。探索建立完善注册人监管体系,明确注册人、受托生产企业等主体之间的法律关系,在责任清晰、风险可控的基础上,构建注册人全生命周期质量监管制度和体系。探索创新监管方式,完善事中事后监管体系,厘清跨区域监管责任,建立完善的跨区域协同监管机制。深化改革和完善医疗器械审评审批制度,落实注册人主体责任,为全面推进实施注册人管理制度积累经验。 二、基本原则 (一)依法依规推进。贯彻中共中央办公厅国务院办公厅印发《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》精神,依据《医疗器械监督管理
条例》《医疗器械注册管理办法》《体外诊断试剂注册管理办法》《医疗器械生产监督管理办法》等法规,依法依规开展试点工作。 (二)全程风险可控。从配套制度设计到实施全过程,全面开展风险评估,明确企业主体责任,实施全生命周期监管,加强上市许可和事中事后监管衔接,落实风险防控措施。 (三)对接国际规则。主动适应医疗器械产业特点和全球化发展趋势,积极借鉴国际委托生产和上市许可通行规则,制定相应的配套制度。 (四)推进改革创新。通过开展注册人制度试点工作,深化医疗器械审评审批制度改革,着力破解实施中的难题,总结评估试点工作经验,为国家全面推进实施注册人制度提供理论和实践参考。 三、主要内容 医疗器械注册申请人(以下简称申请人)申请并取得医疗器械注册证的,成为注册人。申请人可以委托具备相应生产能力的企业生产样品。注册人可以委托一家或多家符合条件的医疗器械生产企业生产已获证产品,并对产品全生命周期质量承担相应法律责任。 (一)产品注册与生产许可分离。申请人可以委托试点省(自治区、直辖市)区域内具备相应生产能力的企业生产样品。注册人不具备相应生产能力的,可以将已获证产品委托给试点省(自治区、直辖市)区域内具备生产能力的一家或多家受托生产企业生产产品。受托生产企业已取得相应生产资质的,应将受托生产产品信息在生产许可证产品列表中登载;受托生产企业不具备相应生产资质的,可提交注册人持有的医疗器械注册证和委托生产相关资料申请办理生产许可。
上海市医疗器械经营管理实施细则
上海市医疗器械经营质量管理规范实施细则 第一章总则 第一条为加强医疗器械经营质量管理,规范医疗器械经营管理行为,保证医疗器械安全、有效,根据《医疗器械监督管理条例》、《医疗器械经营监督管理办法》、《医疗器械经营质量管理规范》等法规规章规定,结合本市实际,制定本实施细则。 第二条本实施细则是医疗器械经营质量管理的基本要求,适用于本市从事医疗器械经营活动的经营者。 医疗器械经营企业(以下简称企业)应当在医疗器械采购、验收、贮存、销售、运输、售后服务等环节采取有效的质量控制措施,保障经营过程中产品的质量安全。 第三条企业应当按照所经营医疗器械的风险类别实行风险管理,并采取相应的质量管理措施,风险管理措施应当符合国家法规规章规定。 第四条企业应当诚实守信,依法经营,禁止任何虚假、欺骗行为。企业提供的资料应当客观真实、不得隐瞒、编造。无《医疗器械监督管理条例》第六十三条、六十四条、六十五条等相关法律法规禁止从业的情形。 在接受食品药品监督管理部门检查时,应当予以配合。 第二章职责与制度 第五条企业法定代表人或者负责人是医疗器械经营质量的主要责任人,全面负责企业日常管理工作。
第六条企业法定代表人或者负责人应当提供必要的条件,保证质量管理机构或者质量管理人员有效履行职责,确保企业按照本实施细则要求经营医疗器械。 第七条企业质量负责人应当由管理层人员担任,熟悉本企业所经营产品的质量特性及质量管理体系,全面负责企业质量管理工作。企业质量负责人应当独立履行职责,在企业内部对医疗器械质量管理具有裁决权,承担相应的质量管理责任。 第八条企业质量管理机构或者质量管理人员应当履行以下职责: (一)组织制订质量管理制度、岗位职责和岗位操作规程,指导、监督制度的执行,并对执行情况进行检查,发现问题及时纠正和持续改进; (二)负责收集医疗器械经营相关的法律、法规、规章等有关规定,实施动态管理,并建立档案和目录清单; (三)负责指导、督促企业相关部门和岗位人员执行医疗器械的法规规章及本实施细则; (四)负责首营审核,包括产品、供货者、购货者合法性资质的审核,同时应当及时收集、更新所经营产品质量信息,并建立所营产品质量档案和目录清单; (五)负责不合格医疗器械的确认,对医疗器械不合格品的确认应当有书面的意见和签字;对不合格品的处理过程,应当实施有效监督,并有书面处理凭证; (六)负责医疗器械质量投诉和质量事故的调查、处理及报告,报告应当有质量负责人意见和签字; (七)组织相关部门验证、校准相关设施设备,相关记录应当保存留档;
申请医疗器械注册证流程细解
申请医疗器械注册证流程细解
申请医疗器械注册证流程细解 医疗器械注册证(II类,非体外诊断试剂类) 一、非体外诊断医疗器械申请材料目录: 资料编号1、医疗器械注册申请表; 资料编号2、医疗器械生产企业资格证明; 资料编号3、产品技术报告; 资料编号4、安全风险分析报告; 资料编号5、适用的产品标准及说明;(应有检测机构签章) 资料编号6、产品性能自测报告; 资料编号7、有承检资质的医疗器械检测机构出具的产品注册检测报告;(原件) 资料编号8、医疗器械临床试验资料;(原件,具体提交方式见《注册管理办法》附件12) 资料编号9、医疗器械说明书;
资料编号10、产品生产质量体系考核(认证)的有效证明文件;(原件) 资料编号11、所提交材料真实性的自我保证声明。 二、非体外诊断医疗器械申请材料要求: (一)申报资料的一般要求: 1、格式要求:(1)申请材料的同一项目的填写应一致;(2)申请材料应使用A4规格纸张打印;(3)申请材料应清晰、整洁,每份申请材料均应装订并加盖企业公章,并按照申请材料目录的顺序装订成册;(4)在每项文件的第一页作一标签,或用带标签的隔页纸分隔,并标明项目编号;(5)用档案袋将报送的材料装好,档案袋需使用封面(格式见“档案袋封面格式.doc”),在袋面标明生产企业名称、地址、产品名称、联系人及电话,并加注申请材料审核的医疗器械注册申请事务人员姓名(需亲笔签名),联系方式,如是医疗器械注册专员请提供姓名(需亲笔签名)、联系方式及备案凭证号。
2、医疗器械注册申请表、产品标准一式两份,其他资料各一份。(附件1~附件5另附,无需与整套申请材料一起装订) 3、各项(上市批件、标准、检测报告、说明书)申报资料中的产品名称应与申请表中填写的产品名称实质性内容相对应。若有商品名,应标注商品名。申报资料应当使用中文,根据外文资料翻译的申报资料,应当同时提供原文。 4、申报资料受理后,企业不得自行补充申请,但属于《医疗器械注册管理办法》第三十八条规定情形的,可以补充申请。 5、生产企业在提交注册申报资料时,应同时提交医疗器械注册申请表、产品注册标准及备案说明书、标签和包装标识的电子文本(Word格式,具体要求见《关于调整医疗器械说明书备案内容的通知》(食药监办[2008]125号),其内容须与纸质文件的内容相一致。电子文本可通过移动存储设备(U盘或光盘)形式提交。 6、办理医疗器械注册申请事务的人员应当受生产企业委托,应提交生产企业负责人身份证
上海医疗器械公司大全
会员号单位名称会员号单位名称 001-A 医疗器械(集团)公司 001-B 医疗器械股份 002 医疗器械股份医疗设备厂 003 医疗器械股份齿科医械厂 004 医疗器械股份齿科材料厂 005 医疗器械股份医用光学仪器厂 006 医疗器械股份医疗器械五厂 007 市医疗器械批发部 008 医疗器械(集团)手术器械厂 009 医疗器械(集团)卫生材料厂010 医疗器械高技术公司 012 窥镜维修站 013 医疗器械厂 014 华辰医用仪表 015 华线医用核子仪器 016 医用分析仪器厂 021 天缘医用生物 022 四菱医疗器械厂 023 医用缝合针厂 025 医用恒温设备 026 医疗器械(集团)医用吸引器厂 028 强生 029 西门子医疗器械 030 阿洛卡医用仪器 031 光电医用电子仪器 032 麦迪逊医疗器械 033 米沙瓦医科工业 034 德尔格医疗器械 035 伯迪克 036 菲曼特医疗器械 037 贺利氏古莎齿科 038 安泰分析仪器 039 胜利医疗器械 041 医疗器械高等专科学校 042 医疗器械研究所 043 医疗器械高等专科学校实验工厂 044 医药职工大学 045 光谊医疗器械厂 046 市药口监督管理局科技情报研究所 049 国药集团医疗器械 050 精密科学仪器 051 实验仪器厂 052 跃进医疗器械厂 053 新亚医用橡胶厂 054 浦东金环医疗用品 055 安亭科学仪器厂 056 三五纸厂 057 三和医疗器械 058 沪通电子 059 跃进医用光学仪器厂 061 康捷医疗用品 062 原子核研究所日环仪器 065 三友外科器材 066 申安医疗器械厂 067 金仪医疗器械厂 069 七0一所园医用氧舱厂 070 市嘉定区医疗设备厂 071 浦卫医疗器械厂 073 光电技术公司 076 市离心机械研究所 077 医科大学仪器厂 079 二医江生物材料 080 工大电子仪器厂 082 交大南洋医疗器械 083 仪表厂有限责任公司 089 公共安全器材厂 090 达华医疗器械 091 爱德沃电器 095 天章记录纸厂 096 康泰医疗器械厂 097 上光学仪器厂 099 昌久医疗器械制造厂 103 中联医疗器械厂 104 爱申医疗器械 105 医达新技术研究所 106 理达仪器厂 107 三科仪器 108 协民医用敷料厂 109 沪江医疗器材 112 安亭电子仪器厂 116 康德莱企业发展(集团) 117 浦东物理光学仪器厂 118 标本模型厂 119 贝琼齿材 121 展旺电子仪器厂 122 新成文教仪器厂
广东省医疗器械注册人生产质量管理体系实施指南(草稿)
附件 广东省医疗器械注册人生产质量管理体系实施指南 (草案) 一、制定依据 为规范医疗器械注册人委托生产质量管理,明确不同主体间的质量管理责任划分,保障委托方和受托方质量管理体系的有效衔接和运行,切实保障医疗器械质量安全,根据《中共中央办公厅国务院办公厅印发关于深化审评审批制度改革鼓励药品医疗器械创新的意见》、《国务院关于印发全面深化中国(广东)自由贸易试验区改革开放方案的通知》、《医疗器械监督管理条例》、《广东省医疗器械注册人制度试点工作实施方案》的要求,结合本省实际,我局制定了《广东省医疗器械注册人生产质量管理体系实施指南(试行)》(以下称:《指南》)。 二、适用范围 医疗器械注册人制度试点是改革完善医疗器械审评审批和注册生产制度的重大创新,建立医疗器械注册人保证医疗器械质量的责任体系、落实企业主体责任是医疗器械注册人制度试点的主要目的之一。 《指南》中的医疗器械注册人是指按照医疗器械注册人制度试点工作要求,委托生产医疗器械样品或产品的企业、机构或科研人员。《指南》中的受托生产企业是指按照医疗器械注册人
制度试点工作要求,接受医疗器械注册人(以下称:注册人)委托生产医疗器械样品或产品的企业(以下称:受托人)。 《指南》旨在为注册人委托生产医疗器械时,对双方在建立、运行、改进医疗器械生产质量管理体系方面提供指导,从而更好地满足《医疗器械生产质量管理规范》及其附录的要求,落实注册人和受托人主体责任,确保医疗器械产品上市后的安全、有效。 《指南》是在《医疗器械生产质量管理规范》及其附录的基础上,针对注册人及其受托人的特殊管理要求所指定的细化指南性意见。注册人及其受托人应在符合《医疗器械生产质量管理规范》及其附录要求的基础上,同时符合医疗器械注册人制度试点和医疗器械委托生产质量协议的相关要求。 《指南》同样适用于医疗器械监管部门在对注册人及其受托人实施医疗器械生产质量管理体系现场核查时参考使用,指导和规范医疗器械监管人员开展注册人委托生产过程的监督检查工作。 三、要求 1义务与责任 1.1注册人 注册人负责医疗器械全链条和全生命周期管理,对医疗器械设计开发、临床试验、生产制造、销售配送、售后服务、产品召回、不良事件报告等承担全部法律责任,确保提交的研究资料
山东医疗器械注册人制度下的生产质量管理体系
医疗器械注册人制度下的生产质量管理体系 医疗器械注册人制度下的生产质量管理体系应符合《医疗器械注册管理办法》、《体外诊断试剂注册管理办法》、《医疗器械生产质量管理规范》及其附录。 医疗器械注册人制度下的生产质量管理体系应结合注册申报资料,重点制定与产品研制、生产有关的“设计开发”、“采购”、“生产管理”、“质量控制”等相关的文件资料。 医疗器械注册申请人应当建立与产品实现过程相适应的质量管理体系并保持有效运行,负责医疗器械全生命周期管理,确保设计开发、生产等过程数据真实可靠、完整、可追溯,并与注册申报资料一致。 医疗器械注册人制度下的生产质量管理体系建设重点 (一)机构与人员 应配备与产品要求相适应的医疗器械设计研发人员,人员应具有与所开发产品相适宜的教育背景、专业知识、工作技能。 (二)厂房、设施、设备 1.产品研发应当在适宜的厂房与设施进行,用于注册检验和临床试验样品生产的厂房与设施,应当满足研发与生产产品的质量控制要求。 2.应当配备与研究项目相适应的场所、设备和仪器,包括用
于注册申报检验和临床试验的样品试制的生产和检测设备,设备能力应当能满足样品试制要求。 3.申报注册质量管理体系现场核查的生产地址应当保留用于注册检验和临床试验的样品试制的厂房与设施,并满足试制量、型号规格的要求。 (三)文件管理 1.对于委托生产的产品,申请人应当确保受托方的文件符合委托方质量管理体系相关要求,并有效控制,包括移交的全部研发资料和技术文件。 2.设计开发原始资料应当纳入文件管理。申请人应当保留产品研制或技术转让后验证的研究数据,确保数据的真实性、完整性和可追溯性;除直接输出的试验数据外,还应当保留研制过程中的辅助记录,如主要物料领用记录、仪器设备使用记录、称量记录、配制记录等,增加研发过程的可追溯性。 (四)设计开发 医疗器械设计开发文档应当源于设计开发评审、验证、确认和设计转换活动的相关文件,包含设计开发程序、开发计划及建立的记录,应当保证对历次设计开发最终输出过程及其相关活动的可追溯性。 1.设计开发输入的依据,应当包括法律法规、国家标准、行—2 —
二类医疗器械注册申请表范本.doc
二类医疗器械注册申请表范本 范本1 受理流水号: 批准流水号; 江苏省第二类医疗器械 注册申请表 产品通用名: 产品商品名: 申请企业:盖章 江苏省食品药品监督管理局制 2014 填写说明
一、本表适用于江苏省第二类医疗器械注册事项的申请。 二、本表由企业填写,需打印。内容应完整、真实。申请企业相关信息应与有效《医疗器械生产企业许可证》及营业执照载明的内容一致。申报产品名称、型号规格原则上应与所提交的注册产品标准或采标声明、注册检测报告、临床试验报告、产品使用说明书一致。产品主要结构和性能、预期用途应与申报材料中的有关内容相一致。 三、根据申请注册种类,在本表“注册形式”栏中相关类型前方框内用“√”做标记。按照规定报送的相关申请材料,在本表“注册申请材料”的名称前方框内用“√”做标记。 四、注册申请表及产品使用说明书应有企业法人代表或法定授权人签字并加盖企业印章;其余每份注册申报资料均应加盖企业印章,必要时加盖骑缝章。五、本表与申报材料一并装订成A4幅面,每注册单元提交一式一份。提交资料中未特别注明的,均应提交原件。 因故不能提交的,应有书面说明。资料详细要求见注册申请材料要求。
产品名称 商品名称 规格型号 注册形式 □ 产品首次注册□ 原注册证有效期届满重新注册 □ 型号规格变化重新注册□ 产品标准内容变化重新注册 □ 生产地址变更重新注册□ 性能、结构及组成变化重新注册 □ 适用范围变化重新注册□ 产品管理类别变化重新注册 生产企业 许可证号 注册地址 邮政编码 生产地址 1 邮政编码 生产地址 2 邮政编码 生产地址 3
邮政编码 法人代表 签字: 传真 联系人 联系电话 手机号码 电子信箱 原注册证号 主要 结构 性能 产品 预期 用途 注册申请材料 □1.医疗器械生产企业许可证正、副本
医疗器械注册管理规定日起施行
医疗器械注册管理规定 日起施行 Document serial number【UU89WT-UU98YT-UU8CB-UUUT-UUT108】
国家食品药品监督管理总局令 第 4 号 《医疗器械注册管理办法》已于2014年6月27日经国家食品药品监督管理总局局务会议审议通过,现予公布,自2014年10月1日起施行。 局长张勇 2014年7月30日 医疗器械注册管理办法 第一章总则 第一条为规范医疗器械的注册与备案管理,保证医疗器械的安全、有效,根据《医疗器械监督管理条例》,制定本办法。 第二条在中华人民共和国境内销售、使用的医疗器械,应当按照本办法的规定申请注册或者办理备案。 第三条医疗器械注册是食品药品监督管理部门根据医疗器械注册申请人的申请,依照法定程序,对其拟上市医疗器械的安全性、有效性研究及其结果进行系统评价,以决定是否同意其申请的过程。 医疗器械备案是医疗器械备案人向食品药品监督管理部门提交备案资料,食品药品监督管理部门对提交的备案资料存档备查。
第四条医疗器械注册与备案应当遵循公开、公平、公正的原则。 第五条第一类医疗器械实行备案管理。第二类、第三类医疗器械实行注册管理。 境内第一类医疗器械备案,备案人向设区的市级食品药品监督管理部门提交备案资料。 境内第二类医疗器械由省、自治区、直辖市食品药品监督管理部门审查,批准后发给医疗器械注册证。 境内第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。 进口第一类医疗器械备案,备案人向国家食品药品监督管理总局提交备案资料。 进口第二类、第三类医疗器械由国家食品药品监督管理总局审查,批准后发给医疗器械注册证。 香港、澳门、台湾地区医疗器械的注册、备案,参照进口医疗器械办理。 第六条医疗器械注册人、备案人以自己名义把产品推向市场,对产品负法律责任。