动物细胞蛋白质的提取和含量的测定
0.0
0.1
0.2
0.3
0.4
0.5
0.0
0.1
0.2
0.3
0.4
0.5
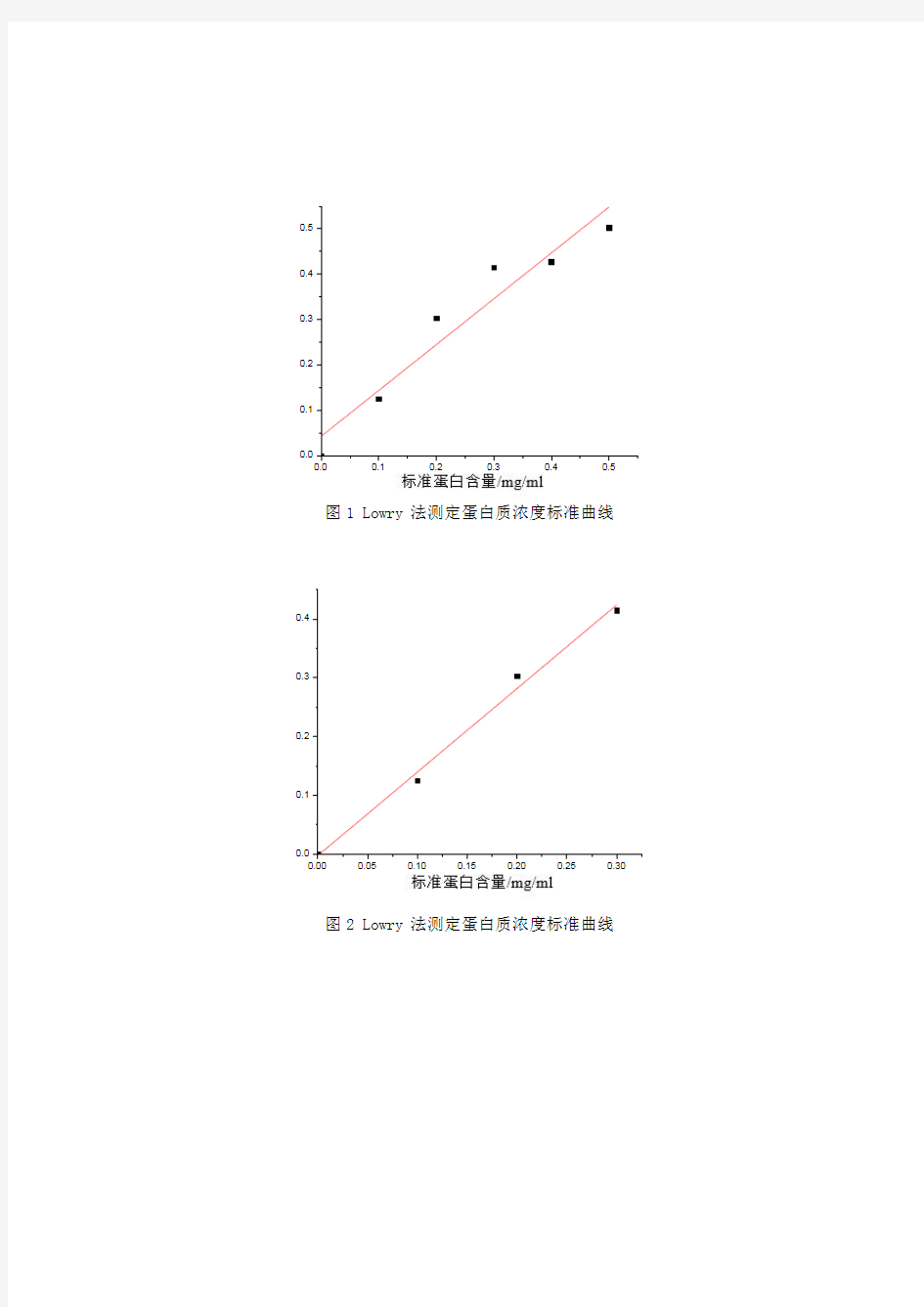
图1 Lowry 法测定蛋白质浓度标准曲线
0.00
0.050.10
0.15
0.20
0.250.30
0.00.1
0.2
0.3
0.4
图2 Lowry 法测定蛋白质浓度标准曲线
0.00.10.20.30.40.5
0.0
0.1
0.2
0.3
0.4
0.5
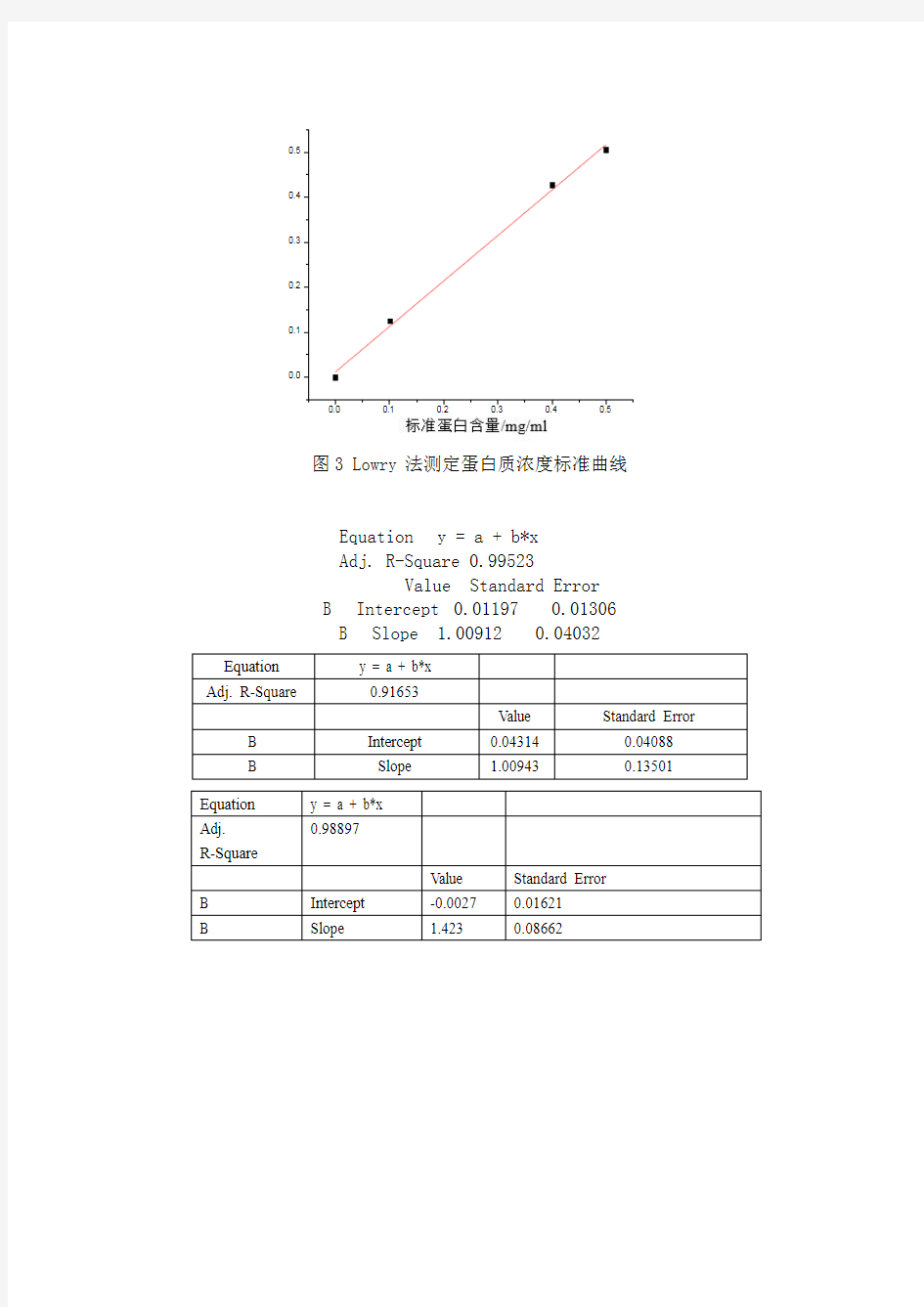
图3 Lowry 法测定蛋白质浓度标准曲线
Equation y = a + b*x Adj. R-Square 0.99523 Value Standard Error B Intercept 0.01197 0.01306 B Slope 1.00912 0.04032
Equation y = a + b*x Adj.
R-Square 0.98897
Value Standard Error B Intercept -0.0027 0.01621 B
Slope
1.423
0.08662
Equation
y = a + b*x Adj. R-Square
0.91653
Value Standard Error B Intercept 0.04314 0.04088 B Slope
1.00943 0.13501
0.0
0.1
0.2
0.3
0.4
0.5
0.00.10.20.3
0.4
0.5
B
图4 考马斯亮蓝法测定蛋白质浓度标准曲线
0.0
0.1
0.2
0.3
0.4
0.0
0.1
0.2
0.3
0.4
0.5
图5 考马斯亮蓝法测定蛋白质浓度标准曲线
Equation y = a + b*x Adj. R-Square
0.99474
Value Standard Error
B Intercept 0.0126 0.0099 B Slope 1.112 0.0404
Equation
y = a + b*x
Adj. R-Square
0.97709
Value Standard Error B Intercept 0.02719 0.02074 B
Slope
1.00257
0.06849
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
图6 紫外吸收法测定蛋白质浓度标准曲线
Equation y = a + b*x Adj. R-Square
0.98185
Value Standard Error B Intercept 0.00414
0.01353 B
Slope
0.57557
0.03493
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
图6 紫外吸收法测定蛋白质浓度标准曲线
Equation
y = a + b*x
Adj. R-Square
0.99513
Value Standard Error B Intercept 0.00181 0.00761 B
Slope
0.56623
0.01979
细胞组织裂解(提蛋白)方法
第一种方法 蛋白匀浆缓冲液: 50 mM Tris-HCl (pH7.5) 150 mM NaCl 5 mM EDTA 1 % NP-40 1 mM PMSF 操作步骤 直接用这个进行组织匀浆 然后于10000 rpm 离心20分钟收集上清 作SDS-PAGE电泳从染色的情况来看所得到的总蛋白纯度都不错条带比较清晰。我是作的小鼠八种常规组织也包括肾脏在内。 将抽提好的蛋白分装后直接放在负八十保存。上样大概3~8ul每孔都可以的具体看个人需要。70v浓缩,150v分离 第二种方法 裂解液配方: 尿素:8M CHAPS:4% DTT:65mM(现加)PMSF:1mM(现加)MiliQ 水操作步骤: 取300mg样品在液氮环境下研磨, 加入1ml裂解液混匀, 室温静置1小时(实验证明改为匀浆更好,蛋白降解轻), 15000g离心1小时, 取上清测定蛋白质浓度。 在裂解液中加IPG buffer有两个作用:1,增强蛋白质的溶解性2,可以结合核酸,在沉淀的时候予以
除去。建议最好加,浓度从0.5-2%不等(这取决于您样品蛋白质的难溶程度)。IPG buffer 是载体两性电解质,IPG buffer另外一个作用是促进蛋白质溶解的,所以IEF前一定要加,裂解液中没有IPG buffer可以么? (可以的,最后上胶条的时候可以再加) 不过,我觉得液氮研磨以后,最好是粗离心一下,把一些结缔组织给离心掉(150g既可)然后再加裂解液。裂解液加1ML应该没什么问题,主要取决你以后蛋白的浓度。用这个方法最好经丙酮沉淀一次比较好) 第三种方法 建议最好加完裂解液后再用超声波破碎,我做肝脏蛋白提取时一般采取400W工作10秒,间歇10秒,次数15次,一次结束后大约30分钟再重复上述步骤一次(因为不同的样品用一根超声柱子,可能会导致污染,所以一定要用双蒸水冲洗干净,再用70%乙醇洗一下,再用水洗一次),另外样品超声时要置于冰浴中,以免产热 做过的是用10ml蛋白裂解液来匀浆3克的组织,最终用考马斯亮兰染色法测得的蛋白浓度大概为30~50 ug/ul 测595 OD值时,设定一个只用水的空白,把所有测得的值都减去这个空白。再用减后的值做标准曲线。查得未知蛋白浓度时,也要用减后的值在标准曲线上查得。 培养细胞 细胞培养于100 mL/L FCS+RPMI1640培养基中,待生长至对数生长期密度约为80%时用细胞括括下细胞离心收集.按细胞/裂解液体积比1:10加入细胞裂解液(7 mol/L尿素,2 mol/L硫
蛋白质的提取与检测
蛋白质的提取与检测
蛋白质的提取与检测 第一节细胞总蛋白的提取及含量测定 【基本原理】 蛋白质含量测定法是生物化学研究中最常用、最基本的分析方法之一。目前常用的有四种经典的方法,即定氮法、双缩脲法(Biuret法)、Folin-酚试剂法(Lowry法)和紫外吸收法。另外还有两种近年普遍使用起来的测定法,即考马斯亮蓝法(Bradford法)与二辛可宁酸法(BCA法)。值得注意的是,上述方法并不能在任何条件下适用于任何形式的蛋白质,因为一种蛋白质溶液用这几种方法测定有可能得出不同的结果。每种测定法都不是完美无缺的,都有其优缺点。在选择方法时应考虑:①实验对测定所要求的灵敏度和精确度;②蛋白质的性质;③溶液中存在的干扰物质;④测定所要花费的时间。 Lowry法:蛋白质与碱性铜溶液中的二价铜离子络和使得肽键伸展,从而使暴露出的酪氨酸和色氨酸在碱性铜条件下与磷钼钨酸反应并产生深蓝色,在750nm有最大光吸收值。在一定浓度范围内,反应液颜色的深浅与蛋白质中的酪氨酸和色氨酸的含量成正比,由于各种蛋白质中的酪氨酸和色氨酸的含量各不相同,因此在测定时需使用同种蛋白质作标准。 Bradford法:蛋白质与染料考马斯亮蓝G-250结合,使得染料最大吸收峰从465nm变为595nm,溶液的颜色由棕黑色变为蓝色。在一定的线性范围内,反应液595nm处吸光度的变化量与反应蛋白量成正比,测定595nm处吸光度的增加即可进行蛋白定量。
BCA (Bicinchoninic acid)法:二价 铜离子在碱性 的条件下,可以 被蛋白质还原 成一价铜离子 (Biuret reaction)并与 BCA相互作用 产生敏感的颜 色反应。两分子 的BCA螯合一 个铜离子,形成 紫色的反应复 合物。该水溶性 的复合物在 562nm处显示 强烈的吸光性, 吸光度和蛋白 浓度在广泛范 围内有良好的 线性关 0.118 0.05 0.154 0.1 0.213 0.2 0.283 0.3 0.329 0.4 0.404 0.5 第二节SDS-PAGE电泳 【基本原理】
蛋白提取步骤
提蛋白及WB的步骤 整个过程细胞或蛋白都必须放冰上 准备工作:前一天准备:借钥匙、检查细胞裂解液相关试剂是否充足、 当天准备:洗玻璃板、开紫外,冰块、碎冰、标记离心管及离心管、细胞刮板、 开低温高速离心机 细胞裂解液配制: 1ml cell lysis 10ul NP-40(离心机后) 25ul 焦磷酸钠 40ul NaF 1ul β-甘油磷酸 2 ul Na3VO4 1ul 蛋白酶抑制剂(用完即放回冰箱) 10ul PMSF(最后加,随加随用,有毒) 细胞裂解和收集 1.观察细胞状态,并准备提蛋白:吸走培养基、用PBS洗细胞两次(倾斜贴壁加PBS,左 右轻轻摇),倾斜贴壁吸走PBS。 2.将细胞盘拿到外间冰上,加裂解液(体积网上推荐:一般106加),冰上静置1-2min。 3.刮细胞:细胞刮板每次用之前拿水涮一涮,甩干,然后从中间到外面打圈刮,再从下往 上,从上往下全面刮,(刮的时候要迅速),最后用枪吸取裂解液至离心管。 细胞破碎 4.超声波破碎细胞:准备三个小烧杯,加满冰块,三个小夹子。(超声波破碎仪的铁棒不 要碰到离心管的壁和底部) 超声波设置: 工作功率5% 工作时间3min 开机时间15s,关机时间30s 温度0度, 报警温度1度 5.4℃、13000r/min,离心15min。(离心机用后一直保持打开的状态,) 蛋白保存 6.蛋白保存及分装:吸上清液至离心管,涡旋、吸一半至另一个管中,涡旋。-80℃保存。注意事项:PMSF一定要现用现加,PMSF在水溶液中不稳定,30min内就会降解一半。样品处理超过1h,补加一次。
BCA测定蛋白浓度 1、测空白板,选差异较小的孔。 BCA测定法波长570,Bracford:595 2、标准蛋白的稀释(5mg/ul):分装1ml PBS来使用,稀释标准蛋白至ul。 取5ul标准蛋白+45ul PBS 3、加样: 浓度0 BSA(ul)0481216 PBS(ul)20161284 4、样品蛋白稀释20倍:2ul蛋白+18ul PBS 5、配BCA:每个孔200ul,一个样品2个重复,A:B=50:1,根据样品数来计算BCA的用量, 一般防止损耗,多算一个样。 6、加BCA:悬空加、37℃孵育30 min。(手不能碰板的底部,影响测定结果) 7、计时30min 8、配分离胶,灌胶,加水封。 9、计时20min。 10、测蛋白:先振板、再设置参数。 11、计算上样体积: 12、配浓缩胶,灌胶,加梳子,等1-2min,出现缩胶的地方补上。 13、打开干式加热器 10、蛋白质上样:根据计算结果乘以稀释倍数,算出蛋白浓度,以体积最大的为准,其他用 水补足。(最大上样量为20ul,除去buffer的体积,蛋白质最大上样体积为16ul) 标记离心管 加样顺序:水、buffer(1/4)、蛋白、离心-涡旋-离心。100℃变性10min(迅速,确保变性程度一致) 11、计时10min ,变性10min。 12、安装电泳槽: 电泳液(1X):配制:90ml(10X)+810ml水(只能用一次) 液面刚覆盖胶,不能有气泡。 13、上样:maker 上样量为3ul。依次上样(吸样品时最好一次吸完)枪头不能撬板,笔直打进去 14、电泳 分离胶100V,20min左右、条带都跑开即可。 浓缩胶145V 1h 有等紫色部分跑到底结束。 转膜 准备工作:转移缓冲液(只能用2次)、三层滤纸、海绵、PVDF膜 PVDF膜用之前用甲醇浸泡数秒。 用海绵和滤纸之前先浸湿。 转膜:负极(黑板)海绵-三层滤纸-胶-PVDF膜-三层滤纸-海绵-正极(白板)。 三层滤纸和膜都需要赶气泡。
细胞的总蛋白质提取全过程及经验
细胞的总蛋白质提取全过程及经验 如果你要自己裂解细胞的时候,需要注意的事项:师兄给你的细胞要赶紧裂解,当然了,你还要看是带瓶子给你还是消化好给你的。一般我使用的时候,是将细胞统一从几个培养瓶中消化收集起来,然后赶紧4度下离心,用预冷的PBS 或者生理盐水洗涤细胞。也有人喜欢在培养瓶中直接加裂解液,这个一般都是做western什么用的,需要的蛋白量不是很多,而且大部分时候都是要有活性的蛋白的。一般1000rpm足够离心了,转速太快,细胞会破碎,然后会损失蛋白。PBS洗涤的最后一遍,记得将细胞转移到超声管中,也许是由于普通的10mL离心管有可能承受不了超声的能量吧,我们师兄师姐一般都用超声管来自装细胞,然后加入裂解液直接超声。3s每次,间歇3s60次,400W,重复工作复位2次,总共超声180次就差不多了。裂解后的细胞要赶紧在2500g下离心30-60min,超声管是没有盖子的,可以直接在上面加一层封口膜离心。在离心的时候,去配制沉淀液,丙酮:无水乙醇:冰乙酸=50:50:0.1,体积比。一般我加的是裂解液体积的5倍。沉淀液要预冷,我喜欢将沉淀液放在-20度下。细胞离心后,上清液加入到沉淀液中,就会看到比较多的白色沉淀出现,-20度沉淀>2h,然后就可以高速沉淀下蛋白了。这里我们用的体积比较大,一般的离心管不能用,要用beckman专用的那种50mL离心管,25000g,15-30min。Beckman离心管比较大,底部是平的,所以蛋白在底部并不是很牢固,在弃去上清液的时候,一定要注意用枪头缓慢的吸出!然后用5mL做有的丙酮洗涤一遍,再用75%酒精将蛋白转移到4mL的离心管中,当然,如果你的蛋白很少,1.5mL的EPtube也可以使用。之所以用4mL是因为我每次提取的蛋白量大概>10mg,而冻干后的蛋白复溶后测定浓度,一般要求在5-6mg/mL之间,所以buffer的体积也会比较的大。这里蛋白在离心的时候,是片状的,一定要慢慢转移,防止损失,不行就多次转移,每次都将离心管离心去上清。转移后的蛋白离心除去酒精,这时候还是会有部分水分的,就在冻干机里面冻干,尽量在室温下操作。冻干后的蛋白质就可以保存起来了。然后在使用之前,测定蛋白质的浓度,如果你用了8M尿素溶解,在酶解的时候,就需要稀释到<2M,所以蛋白初始浓度不能太低。在蛋白复溶的时候,尽量在4度操作,然后一定要慢慢的加buffer,加多了就不好了,只要蛋白能完全溶解就好。完全溶解的蛋白,会有些粘稠,但是绝对不会有不溶物产生,如果产生了不溶物,就说明需要加buffer,一般复溶这一步要进行一个下午最少!
总蛋白的提取
1.单层贴壁细胞总蛋白的提取: (1)倒掉培养液,并将瓶倒扣在吸水纸上使吸水纸吸干培养液(或将瓶直立放置一会儿使残余培养液流到瓶底然后再用移液器将其吸走)。 (2)每瓶细胞加3ml 4℃预冷的PBS(0.01M pH7.2~7.3)。平放轻轻摇动1min洗涤细胞,然后弃去洗液。重复以上操作两次,共洗细胞三次以洗去培养液。将PBS弃净后把培养瓶置于冰上。 (3)按1ml裂解液加10 μl PMSF(100mM),摇匀置于冰上。(PMSF要摇匀至无结晶时才可与裂解液混合。) (4 )每瓶细胞加400 μl含PMSF的裂解液,于冰上裂解30min,为使细胞充分裂解培养瓶要经常来回摇动。 (5 )裂解完后,用干净的刮棒将细胞刮于培养瓶的一侧(动作要快),然后用枪将细胞碎片和裂解液移至1.5ml离心管中。(整个操作尽量在冰上进行。) (6 )于4℃下12000rpm离心5min。(提前开离心机预冷) (7 )将离心后的上清分装转移到0.5ml的离心管中放于-20℃保存。 2. 组织中总蛋白的提取: (1 )将少量组织块置于1~2ml匀浆器中球状部位,用干净的剪刀将组织块尽量剪碎。 (2)加400 μL单去污剂裂解液裂(含PMSF)于匀浆器中,进行匀浆。然后置于冰上。 (3 )几分钟后再碾一会儿再置于冰上,要重复碾几次使组织尽量碾碎。 (4)裂解30 min后,即可用移液器将裂解液移至1.5ml离心管中,然后在4℃下12000rpm离心5min,取上清分装于0.5ml离心管中并置于-20℃保存。 3. 加药物处理的贴壁细胞总蛋白的提取: 由于受药物的影响,一些细胞脱落下来,所以除按(一)操作外还应收集培养液中的细胞。以下是培养液中细胞总蛋白的提取: (1 )将培养液倒至15ml离心管中,于2500rpm离心5min。 (2 )弃上清,加入4ml PBS并用枪轻轻吹打洗涤,然后2500rpm离心5min。弃上清后用PBS重复洗涤一次。 (3)用枪洗干上清后,加100 μL裂解液(含PMSF)冰上裂解30min,裂解过程中要经常弹一弹以使细胞充分裂解。 (4)将裂解液与培养瓶中裂解液混在一起4℃、12000rpm离心5min,取上清分装于0.5ml离心管中并置于-20℃保存。 含量测定
蛋白质提取及纯化
蛋白质提取及纯化 提取蛋白质的当天早晨去后把高速离心机和超高速离心机都打开冷却 1、前一天晚上用Resuspension Buffer重悬4L菌体,然后离心于4C保存,第 二天使用。 2、用少量预冷的Resuspension Buffer重悬细菌,1 protease inhibitor tablets(EDTA Free),1mM PMSF, 然后用玻璃Homogenizer做均一化处理,将总体积调至80ml; 3、High Pressure Homogenizer破壁,特别注意样品一定要在不加压力的情况 下运行一个循环(2min);然后1200bar,6min三个循环,整个过程冰水冷却; 4、DNaseI处理:加入2.5mg DNaseI,10mM MgCl2, 室温处理30min; 5、 11.000rpm,4℃,15min; then 11.000rpm,4℃,15min; 6、 1mM PMSF, 45.000rpm,4℃,90min; 7、用Resuspension Buffer洗两次以除去可溶性的蛋白质,然后预热分光光度 计; 8、用3-4ml Binding Buffer重悬Membrane pellets,动作一定要轻缓,重悬 后的总体积不超过8ml,取出300ul测定OD800和OD850(以OD850为准),测定时候是逐步稀释,每次吸光值小于1; 9、调整OD850≤30-50,在缓慢搅拌(速度一定要慢)的情况下逐滴加入30%的 LDAO使其终浓度达到0.5%,1mM PMSF,26℃黑暗条件下重悬1h,期间注意观察颜色变化; 10、45.000rpm, 4℃, 30min,注意观察颜色的变化以及沉淀是否发生明显的变化。 Charge and Equilibrate Resin (1)用蒸馏水冲洗柱子以除去20%酒精,注意不要用buffer,1ml/min,至紫外 吸收和电导稳定; (2)用0.1M NiSO4 Charge Resin,1ml/min,10倍柱体积,尽量使得紫外吸收 和电导稳定; (3)用蒸馏水冲洗,1ml/min,至紫外吸收和电导稳定;
细胞总蛋白提取方法
细胞总蛋白提取方法 一、溶液配制 1.100mM NaF (NaF MW=41.99) 10ml 称取NaF 41.99mg,超纯水8ml溶解后定容至10ml。 2.100 mM PMSF 3.RIPA溶液100ml 成分分子量终浓度 Tris 121.14 0.6 g 5.0 mM (pH 7.4) NaCl 58.44 0.87 g 150 mM EDTA 372.24 37.22 mg 1 mM NP-40 1 ml 1% SDS 0.1 g 0.1% 脱氧胆酸钠0.5 g 0.5% 溶解于80 ml超纯水中,待溶解后加入HCl调pH值至7.4,定容至100 ml。 ※使用前加入 成分储存浓度加入量终浓度 PMSF 100 mM 10μl/1ml 1 mM NaF 100 mM 10μl/1ml 1 mM Aprotinin 10μg/μl0.2μl/1ml 2μg/μl Leupetin 10μg/μl0.2μl/1ml 2μg/μl 二、方法 1.细胞收取 1)细胞传代至60mm培养皿中,待细胞融合约100%,收集细胞 2)弃培养基,加入PBS 2ml清洗培养皿一次,弃PBS。 3)加入0.05%胰酶2ml,37℃温育1min。 4)待细胞变形后,将细胞吹下转移至一个1.5ml ependorf 管中,10,000rpm×3min,4℃ 5)弃上清,1ml预冷的PBS清洗沉淀,10,000rpm×
6)重复PBS清洗一次 7)弃上清,-80℃冻存待裂解 2.蛋白提取 1)向细胞沉淀中加入200μl RIPA缓冲液(含1mM PMS 1mM NaF、2μg/ml Aprotinin、2μg/ml Leupetin) 后冰上放置40mins 2)10,000rpm×15min,4℃ 将上清转移至一个新的ependorf 管中(约250μl)
蛋白质提取综合性实验
生物化学综合性实验 蛋白质的提取(沉淀法)和定量分析之 鸡蛋中卵清蛋白的提取和定量测定 一、实验目的 研究盐析沉淀和等电点沉淀法的基本原理和技术。 一、二、实验原理 1、沉淀法粗分离蛋白质[1][2] 沉淀法是分离纯化生物大分子物质常用的一种经典方法,可分盐析法、等电点沉淀法和有机溶剂沉淀法等。 蛋白质分子表面含有带电荷的基团,这些基团与水分子有较大的亲和力,故蛋白质在水溶液中能形成水化膜,增加了蛋白质水溶液和稳定性。如果在蛋白质溶液中加入大量中性盐,导致蛋白质分子表面电荷被中和,水化膜被破坏,最终引起蛋白质分子间相互聚集并从溶液中析出,这就是盐析作用。 由于各种蛋白质分子表面的极性基团所带电荷数目不同,它们在蛋白质表面上的分布情况也不一样,因此,将不同蛋白质盐析出来所需要的盐浓度也各异,盐析法就是通过控制盐的浓度,使蛋白质混合液中的各个成分分步盐析出来,达到粗分离蛋白质的目的。 盐析法是1878年Hammarster首次使用的,可用作盐析的中性盐有过硫酸钠、氯化钠、磷酸钠、硫酸铵等,其中应用最广的是硫酸铵,硫酸铵在水中溶解度大,25℃可达4.1mol/L的浓度,化学性质稳定,溶解度的温度系数变化较小,价廉易得;分段效果较其他盐好,性质温和,即使浓度很高时也不会影响蛋白质的生物学活性。 鸡蛋清的主要成分是球蛋白和白蛋白(卵清蛋白),球蛋白可在半饱和硫酸铵溶液中析出,而清蛋白则在饱和硫酸铵溶液中才能析出。 蛋白质的盐析作用是可逆过程,由盐析获得的蛋白质沉淀,当降低其盐类浓度时,又能再溶解,因而可初步纯化蛋白质。 等电点沉淀法是利用蛋白质在其等电点时溶解度最小来分离具有不同等电点蛋白质的方法。蛋白质是两性电解质,蛋白质分子的电荷性质和数量因PH不同而变化,蛋白质处于等电点时,其净电荷为零,由于相邻蛋白质分子之间没有静电斥力而趋于聚集沉淀,因此,在其他条件相同时,它的溶解度达到最低点。 卵清蛋白的等电点为4.6-4.8,而球蛋的等电点是5.1。 2、蛋白质的测定 根据蛋白质的物理化学性质,测定蛋白质的方法有凯氏定氮法、紫外吸收法、Folin-酚法、考马斯亮蓝G-250染色法等。 由于蛋白质分子中酪氨酸和色氨酸残基的苯环含有共轭双键,因此蛋白质具有吸收紫外线的性质,吸收高峰在280nm波长处。在此波长范围内,蛋白质溶液的光吸收值(A280)与其含量呈正比关系,可用作定量测定。 由于核酸在280波长处也有光吸收,对蛋白质的测定有干扰作用,但核酸的最大吸收峰在260nm处,如同时测定260nm的光吸收,通过计算可能消除其对蛋白质测定的影响,因此溶液中存在核酸时必须同时测定280nm及260nm之光密度,方可通过计算测得
蛋白质提取方法
蛋白质提取方法-------列举10种方法 一、植物组织蛋白质提取方法(summer) 1、根据样品重量(1g样品加入3.5ml提取液,可根据材料不同适当加入),准备提取液放在冰上。 2、把样品放在研钵中用液氮研磨,研磨后加入提取液中在冰上静置(3-4 小时)。 3、用离心机离心8000rpm40min4℃或11100rpm20min4℃ 4、提取上清夜,样品制备完成。 蛋白质提取液:300ml 1、1Mtris-HCl(PH8)45ml 2、甘油(Glycerol)75ml 3、聚乙烯吡咯烷酮(Polyvinylpolypyrrordone)6g 这种方法针对SDS-PAGE,垂直板电泳! 二、植物组织蛋白质提取方法(summer) 三氯醋酸—丙酮沉淀法 1、在液氮中研磨叶片 2、加入样品体积3倍的提取液在-20℃的条件下过夜,然后离心(4℃8000rpm以上1小时)弃上清。 3、加入等体积的冰浴丙酮(含0.07%的β-巯基乙醇),摇匀后离心(4℃8000rpm以上1 小时),然后真空干燥沉淀,备用。 4、上样前加入裂解液,室温放置30 分钟,使蛋白充分溶于裂解液中,然后离心(15℃8000rpm 以上1小时或更长时间以没有沉淀为标准),可临时保存在4℃待用。 5、用Brandford法定量蛋白,然后可分装放入-80℃备用。 药品: 提取液:含10%TCA 和0.07%的β-巯基乙醇的丙酮 裂解液:2.7g 尿素0.2gCHAPS 溶于3ml 灭菌的去离子水中(终体积为5ml),使用前再加入1M 的DTT65ul/ml。 这种方法针对双向电泳,杂质少,离子浓度小的特点!当然单向电泳也同样适用,只是电泳的条带会减少! 三、组织:肠黏膜(newinbio) 目的:WESTERN BLOT检测凋亡相关蛋白的表达 应用TRIPURE 提取蛋白质步骤: 含蛋白质上清液中加入异丙醇:(1.5ml每1mlTRIPURE用量)倒转混匀,置室温10min 离心:12000 g,10min,4度,弃上清,加入0.3M盐酸胍/95%乙醇:(2ml每1mlTRIPURE 用量)振荡,置室温20min 离心:7500g,5 min,4 度,弃上清 重复0.3M盐酸胍/95%乙醇步2 次 沉淀中加入100%乙醇2ml 充分振荡混匀,置室温20 min 离心:7500g,5min,4度,弃上清吹干沉淀,1%SDS溶解沉淀 离心:10000g,10min,4度 取上清-20 度保存(或可直接用于WESTERN BLOT) 存在的问题:加入1%SDS 后沉淀不溶解,还是很大的一块,4 度离心后又多了白色沉定,SDS 结晶?测浓度,含量才1mg/ml左右。 解决:提蛋白试剂盒,另外组织大小适中,要碎,立即加2X BUFFER,然后煮5-10分钟,
细胞的总蛋白质提取全过程及经验
细胞的总蛋白质提取全 过程及经验 The manuscript was revised on the evening of 2021
细胞的总蛋白质提取全过程及经验 如果你要自己裂解细胞的时候,需要注意的事项:师兄给你的细胞要赶紧裂解,当然了,你还要看是带瓶子给你还是消化好给你的。一般我使用的时候,是将细胞统一从几个培养瓶中消化收集起来,然后赶紧4 度下离心,用预冷的PBS 或者生理盐水洗涤细胞。也有人喜欢在培养瓶中直接加裂解液,这个一般都是做 western 什么用的,需要的蛋白量不是很多,而且大部分时候都是要有活性的蛋白的。一般1000rpm 足够离心了,转速太快,细胞会破碎,然后会损失蛋白。PBS 洗涤的最后一遍,记得将细胞转移到超声管中,也许是由于普通的10mL 离心管有可能承受不了超声的能量吧,我们师兄师姐一般都用超声管来自装细胞,然后加入裂解液直接超声。3s 每次,间歇3s 60 次,400W,重复工作复位2 次,总共超声180 次就差不多了。裂解后的细胞要赶紧在2500g 下离心30-60min,超声管是没有盖子的,可以直接在上面加一层封口膜离心。在离心的时候,去配制沉淀液,丙酮:无水乙醇:冰乙酸=50:50:,体积比。一般我加的是裂解液体积的5 倍。沉淀液要预冷,我喜欢将沉淀液放在-20 度下。细胞离心后,上清液加入到沉淀液中,就会看到比较多的白色沉淀出现,-20 度沉淀>2h,然后就可以高速沉淀下蛋白了。这里我们用的体积比较大,一般的离心管不能用,要用 beckman 专用的那种50mL 离心管,25000g,15-30min。Beckman 离心管比较大,底部是平的,所以蛋白在底部并不是很牢固,在弃去上清液的时候,一定要注意用枪头缓慢的吸出!然后用5mL 做有的丙酮洗涤一遍,再用75%酒精将蛋白转移到4mL 的离心管中,当然,如果你的蛋白很少,的EP tube 也可以使用。之所以用4mL 是因为我每次提取的蛋白量大概>10mg,而冻干后的蛋白复溶后测定浓度,一般要求在5-6mg/mL 之间,所
蛋白提取实验步骤
蛋白提取实验步骤: 1、细胞总蛋白提取 A、对于悬浮细胞: 离心收集细胞,每106细胞加250 ul RIPA (在使用前数分钟内加入蛋白酶抑制剂),振荡。如果需要提高蛋白浓度,可以适当减少细胞总蛋白提取试剂体积。 B、对于贴壁细胞: a、用TBS冲洗细胞2-3次。最后一次彻底吸干残留液。 b、加入适当体积的 RIPA(使用前数分内加入蛋白酶抑制剂)于培养板、瓶内3-5分钟。期间反复晃动培养板、瓶,使试剂与细胞充分接触。 c、用细胞刮刀将细胞及试剂刮下,收集到1.5ml离心管中。 C、冰浴30min,期间用移液器反复吹打,确保细胞完全裂解。 D、12000g离心5min,收集上清,即为总蛋白溶液。 2、组织蛋白提取: A、组织块用冷TBS洗涤2-3次,去除血污,剪成小块置于匀浆器。加入10倍组织体积本试剂(使用前数分钟内加入蛋白酶抑制剂)冰上彻底匀浆。如果需要提高蛋白浓度,可以适量减少该试剂体积。 B、将匀浆液转移至1.5ml离心管中,振荡。冰浴30min,期间用移液器反复吹打,确保细胞完全裂解。 C、12000g离心5min,收集上清,即为总蛋白溶液。 注意事项 1、组织尽可能新鲜,若不能及时提蛋白,组织标本应保存在-80℃冰箱,并避免反复冻融。 2、全程必须在冰上进行,避免蛋白降解。 3、蛋白酶抑制剂在水溶液中不稳定,需在RIPA使用前数分钟加入蛋白酶抑制剂。
4、裂解过程中如有溶液粘稠现象可用移液器(200μl)反复吹打,或再加入适量裂解液以保证充分裂解。 5、总蛋白溶液不稳定(蛋白酶依旧有活性)可在-80℃短时间保存,建议立即加入蛋白上样缓冲液变性后与-20℃保存,避免反复冻融。
【1】生物样本中蛋白质的提取及测定(分子医学实验)
《分子生物学实验》 实验报告 实验名称:生物样本中蛋白质的提取及测定 姓名:杰 学号:3140104666 组别: 同组同学:唐曦
带教教师:伟俞萍 实验日期:2015年9月15日 目录 1.原理: (3) 1.1生物样本中蛋白质的提取 (3) 1.2生物样本中蛋白质的测定 (3) 1.2.1 Lowry法 (3) 1.2.2 考马斯亮蓝法 (4) 1.2.3 紫外吸收法 (4) 2.操作步骤 (4) 2.1生物样本中蛋白质的提取 (4) 2.2生物样本中蛋白质的测定 (5) 2.2.1 Lowry法 (5) 2.2.2 考马斯亮蓝法 (5) 2.2.3紫外吸收法 (5) 3、实验结果 (6) 3.1 原始数据 (6) 3.1.1 Lowry法 (6) 3.1.2 考马斯亮蓝法 (7) 3.1.3 紫外吸收法 (7)
3.2 数据处理 (8) 3.2.1 Lowry法 (8) 3.2.2 考马斯亮蓝法 (9) 3.2.3 紫外吸收法 (10) 4.讨论: (11) 1.原理: 1.1生物样本中蛋白质的提取 离体不久的组织,在适宜的温度及pH等条件下,可以进行一定程度的物质代谢。因此,在生物化学实验中,常利用离体组织来研究各种物质代谢的途径与酶系作用,也可以从组织中提取各种代谢物质或酶进行研究。但生物组织离体过久,其所含物质的含量和生物活性都将发生变化。例如,组织中的某些酶在久置后会发生变性而失活;有些组织成分如糖原、ATP等,甚至在动物死亡数分钟至十几分钟,其含量即有明显的降低。因此,利用离体组织作代谢研究或作为提取材料时,都必须迅速将它取出,并尽快地进行提取或测定。一般采用断头法处死动物,放出血液,立即取出实验所需的脏器或组织,除去外层的脂肪及结缔组织后,用冰冷的生理盐水洗去血液(必要时可用冰冷的生理盐水灌注脏器以洗去血液),再用滤纸吸干,即可用于实验。取出的脏器或组织,可根据不同的方法制成不同的组织样品。包括组织糜、组织匀浆、组织浸出液。由于动物肝脏细胞比较脆弱,易于破碎,故本实验选用小鼠肝脏细胞作为实验材料,采用匀浆法法将其破碎,然后加入样品提取液使蛋白质溶解,用高速离心法弃去细胞碎片。收集上清液后可进行蛋白质定量分析。 1.2生物样本中蛋白质的测定 1.2.1 Lowry法 1921年,Folin发明了Folin-酚试剂法测定蛋白质的浓度,反应原理是利用蛋白质分子中的酪氨酸和色氨酸残基还原酚试剂(磷钨酸-磷泪酸)生成蓝色
蛋白提取方法
?1 材料与方法 1、1 材料 1、1、1组织与细胞得来源: ?1。1、2 仪器设备?机械组织匀浆器?低温高速离心机(>40,000g) 超速离心机?超生细胞破碎仪 超纯水装置 ??1、1。3 试剂 三氯醋酸(TCA) 丙酮 二硫苏糖醇(DTT) ?尿素?CHAPS?PMSF?EDTA ?乙醇?磷酸 考马斯亮蓝R350 ?抑肽素A?亮肽素 试剂纯度均应就是分析纯或以上。? 1。1、4溶液配制?(1) PBS: ?NaCl8g,KCl 0、2 g,Na2HPO4 1。44 g,KH2PO4,溶于800 ml水中,用HCl调pH至7.4,用纯水定容至1 L; ?(2)EDTA 储存液: 18.61g Na2EDTA?2H2O,溶于70 ml纯水中,用10 mol/L NaOH调节pH值至8.0(约需2 g NaOH颗粒),定容为100 ml、可高压灭菌后分装备用;?(3) 亮肽素储存液(50 μg/ml,100×) 10mg/ml溶于水,-75℃保存;使用时配成50 μg/ml储液,-20℃保存; (4) 抑肽素储存液(70μg/ml,100×) 1 mg/ml溶于甲醇,—75℃保存;使用时配成70 μg/ml储液,-20℃保存; (5) PMSF储存液(10mM,100×): 17.4mgPMSF,溶于1ml异丙醇中,—20℃保存。?DTT储存液(1 M): ?0。31gDTT溶于2 mlH2O中,-20℃保存(DTT或含有DTT得溶液不能进行高压处理,可过滤除菌)。?(7) 裂解液: Lysis buffer A (9M urea,4%w/v CHAPS, 1%w/v DTT, 0.5%CA and a cocktailof proteaseinhibitors)??Lysis buffer B (7 M urea, 2M thiourea,4% w/v CHAPS, 1%w/v DTT,0、5% CA andacocktailofprotease inhibitors) ?Lysisbuffer C 40 mMTris-base (pH 9。5)inultrapure H2O Lysis buffer D (8 Murea, 4%CHAPS, 40mM Tris(base), 40 ml) ?Lysis buffer E ?(5M urea, 2 M thiourea, 2%SB3-10, 2%CHAPS,1% w/v DTT, 0、5% CAandacocktail ofprotease inhibitors) 100μL SDSsample solution (1% w/v SDS, 0、375M Tris-HCl, pH8。8, 50 mM DTT,25%v/vgly ?LysisbufferF? cerol) ●CA、蛋白酶抑制剂混合物与DTT在临用前加入。?蛋白酶抑制剂混合物[3]?成分终浓度?蛋白酶抑制剂混合物 PMSF 35 μg/ml or 1 mM EDTA 0、3mg/ml (1 mM) 抑肽素 0。7 μg/ml?亮肽素 0、5μg/ml? 1.2 方法?1。1.1组织蛋白提取方法 1、2.1。1三氯醋酸/丙酮沉淀法[1]?(1)冰上取材,称湿重,置液氮中冻存或直接进行下一步; (2)在液氮中研碎样品或使用机械匀浆器磨碎组织; ?(3)将粉末悬浮于含DTT(0。2%w/v)得10%三氯醋酸(w/v)得丙酮溶液中; (4)蛋白–20℃沉淀过夜; ?(5)35000×g(6℃)离心30min; ?将沉淀重悬于含0.2%DTT得预冷丙酮中; ?(7)-20℃放置1h; 35000×g(6℃)离心30min; (9)在通风橱中让丙酮充分挥发,得到干燥得沉淀; 15℃,40000×g,离心1hr; (10)在裂解液中重新溶解沉淀(50-100mg组织需要1ml裂解液); ?( ) 11 (12)用Bradford法[2]测定上清得蛋白浓度,分装后置–75℃保存。?1、2、1。2超速离心法?(1)取材; ?(2)用研钵在液氮冷冻条件下将样品
提取蛋白的常规方法
1、原料的选择 早年为了研究的方便,尽量寻找含某种蛋白质丰富的器官从中提取蛋白质。但至目前经 常遇到的多是含量低的器官或组织且量也很小,如下丘脑、松果体、细胞膜或内膜等原材料, - 105 - 蛋白质提取与制备Protein Extraction and Preparation 因而对提取要求更复杂一些。 原料的选择主要依据实验目的定。从工业生产角度考虑,注意选含量高、来源丰富及成 本低的原料。尽量要新鲜原料。但有时这几方面不同时具备。含量丰富但来源困难,或含量 来源均理想,但分离纯化操作繁琐,反而不如含量略低些易于获得纯品者。一般要注意种属 的关系,如鲣的心肌细胞色素C 较马的易结晶,马的血红蛋白较牛的易结晶。要事前调查 制备的难易情况。若利用蛋白质的活性,对原料的种属应几乎无影响。如利用胰蛋白酶水解 蛋白质的活性,用猪或牛胰脏均可。但若研究蛋白质自身的性质及结构时,原料的来源种属 必须一定。研究由于病态引起的特殊蛋白质(本斯.琼斯氏蛋白、贫血血红蛋白)时,不但 使用种属一定的原料,而且要取自同一个体的原料。可能时尽量用全年均可采到的原料。对 动物生理状态间的差异(如饥饿时脂肪和糖类相对减少),采收期及产地等因素也要注意。 2、前处理 a、细胞的破碎 材料选定通常要进行处理。要剔除结缔组织及脂肪组织。如不能立即进行实验,则应冷 冻保存。除了提取及胞细外成分,对细胞内及多细胞生物组织中的蛋白质的分离提取均须先 将细胞破碎,使其充分释放到溶液中。不同生物体或同一生物体不同的组织,其细胞破坏难 易不一,使用方法也不完全相同。如动物胰、肝、脑组织一般较柔软,作普通匀浆器磨研即 可,肌肉及心组织较韧,需预先绞碎再制成匀桨。 ⑴机械方法 主要通过机械切力的作用使组织细胞破坏。常用器械有:①高速组织捣碎机(转速可达 10000rpm,具高速转动的锋利的刀片),宜用于动物内脏组织的破碎;②玻璃匀浆器(用两 个磨砂面相互摩擦,将细胞磨碎),适用于少量材料,也可用不锈钢或硬质塑料等,两面间
蛋白含量测定及western步骤
蛋白的提取和定量 肺组织用预冷1×TBS洗净后,加入含PMSF的RIPA buffer(冰上操作,310ul,决定未来的蛋白浓度和蛋白液体积),50-60mg肺组织砸碎放入1.5ml离心管,冰上孵育1h,10000转4℃离心10min,转上清至新管。裂解液分装后保存于-70℃ 蛋白质定量:BCA蛋白测定法 ①根据样品数量,按50体积BCA试剂A加1体积BCA试剂B(50:1)配制适量BCA工作液,充分混匀。BCA工作液室温24小时内稳定。 ②完全溶解蛋白标准品(BCA试剂盒中,BSA原浓度2mg/mL),稀释到1mg/mL。 ③将标准品按0,2,5,10,15,20,25 ul标准品孔中,加蒸馏水稀释标准品的 ④加样品2uL加到96孔板的样品孔中,加蒸馏水23微升。 ⑤各孔加入200微升BCA工作液,37o C放置30分钟。同时打开酶标仪预热。 注:也可以室温放置2小时,或60o C放置30分钟。BCA法测定蛋白浓度时,吸光度会随着时间的延长不断加深。并且显色反应会因温度升高而加快。如果浓度较低,适量在较高温度孵育,或延长孵育时间。 ⑥测定A570的波长,根据标准曲线计算出蛋白浓度。 ⑦计算调蛋白时所需TBS和RSB的体积(调所有样品浓度至3-5ug/ul): 总体积=蛋白体积*蛋白浓度/3(ul) RSB=1/5*总体积(ul) TBS=总体积-RSB-蛋白体积(ul) 先加RSB(对蛋白有保护作用),后加TBS。最后放于-70℃保存。 Western Blot SDS-PAGE 1. 玻璃板:注意对齐、夹紧,防止漏出,短板朝前。 灌至距绿线1cm左右,用dd水封顶,放置30-40min。状况好时往往能观察到
细胞总蛋白的制备,SDS-PAGE,WesternBlot
医学生物学研究技术与实验 实验报告 实验名称:细胞总蛋白的制备,SDS-PAGE,Western Blot 一、实验目的 1. 掌握SDS直接裂解细胞法提取蛋白质 2. 掌握Lowry法测定蛋白质浓度的原理 3. 掌握SDS-PAGE分离蛋白质的原理和技术 4. 掌握半干转移的操作和Western Blot 的原理和技术 二、实验原理 1. 蛋白质提取常见裂解方法极其原理。 蛋白质的抽提是指破碎过程中,将生物材料在水,缓冲液或稀盐溶液等适当溶剂中浸泡,使胞内的蛋白质等内容物释放到溶剂中。血浆,消化液和分泌液等体液中可溶性蛋白质,可不经抽提,直接进行分离。细胞内一般蛋白质的抽提,应先将细胞膜或细胞壁破碎,然后用适当溶剂将蛋白质溶出,再用离心法除去不溶物,得到出抽提液。膜蛋白的抽提比较复杂。膜蛋白按其所在位置分为外周蛋白和固有蛋白。外周蛋白通过次级键和膜外侧脂质的性头部鳌和在一起,应选则适当离子强度及PH的缓冲液,其中要好友EDTA,将其抽出。固有蛋白嵌和在膜脂质双层中,通过疏水键于膜内侧脂质层的疏水性尾部结合。在抽提固有蛋白时,要减弱器与膜脂的疏水性结合,又要使其保持部分疏水基暴露在外的天然状态,这一过程叫增溶作用。较为理想的增溶剂是去垢剂。目前用的去垢剂分为阴离子型,阳离子型,两性离子型,非离子型。增溶后的膜蛋白抽提剂有较好的均一性,便于进一步纯化。纯化后的膜蛋白,可通过透析法去除去垢剂,进行膜蛋白重组。抽提蛋白质的理想条件是尽可能促进蛋白质在溶剂中溶解,而减弱蛋白水
解酶活力,以减少细胞的自溶过程。主要是通过选择适当PH,温度,或溶剂,以及加适当蛋白水解酶抑制剂。常见裂解方法有:1 低渗裂解,2 冻融法,3 Triton100或者NP-40等非离子去污剂(比较温柔),4 脱氧胆酸钠、SDS、Triton100 (强烈),5 上样缓冲液(含SDS)+细胞沸水浴5 min,6 匀浆器。 2. Lowry法测定蛋白质浓度 1. 蛋白质与碱性铜试剂产生双缩脲反应,形成紫红色蛋白质-铜复合物; 2. 紫红色复合物中的酪氨酸和色氨酸残基还原磷钼酸和磷钨酸,产生深蓝色,呈色强度与蛋白质浓度呈正相关,分光光度计测A750吸光度,做标准曲线 3.SDS-PAGE分离蛋白质 SDS-聚丙烯酰胺凝胶电泳是蛋白分析中最经常使用的一种方法.它是将蛋白样品同离子型去垢剂十二烷基硫酸钠(SDS)及巯基乙醇一起加热,使蛋白变性,多肽链内部及肽键之间的二硫键被还原,肽键被打开,打开的肽键靠疏水作用与SDS结合而带负电荷,电泳时在电场作用下,肽链在凝胶中向正极迁移。不同大小的肽链由于在迁移时受到的阻力不同,在迁移过程中逐渐分开,其相对迁移率与分子量的对数间成线形关系。 4. 非变性聚丙烯酰胺凝胶电泳(Native-PAGE) 非变性聚丙烯酰胺凝胶电泳(Native-PAGE)是在不加入SDS 疏基乙醇等变性剂的条件下,对保持活性的蛋白质进行聚丙烯酰胺凝胶电泳,常用于同工酶的鉴定和提纯。未加SDS的天然聚丙烯酰胺凝胶电泳可以使生物大分子在电泳过程中保持其天然的形状和电荷,它们的分离是依据其电泳迁移率的不同和凝胶的分子筛作用,因而可以得到较高的分辨率,尤其是在电泳分离后仍能保持蛋白质和酶等生物大分子的生物活性,对于生物大分子的鉴定有重要意义,其方法是在凝胶上进行两份相同样品的电泳,电泳后将凝胶切成两半,一半用于活性染色,对某个特定的生物大分子进行鉴定,另一半用于所有样品的染色,以分析样品中各种生物大分子的种类和含量。 5. 不连续聚丙烯酰胺凝胶电泳基本原理 不连续聚丙烯酰胺凝胶电泳包含了两种以上的缓冲液成分、pH值和凝胶孔径,而且在电泳过程中形成的电位梯度亦不均匀。由此产生的浓缩效应、电荷效应和分子筛效应。 (1)浓缩效应样品在电泳开始时,通过浓缩胶被浓缩成高浓度的样品薄层(一般能浓缩几百倍),然后再被分离。当通电后,在样品胶和浓缩胶中,解离度最大的Cl—有效迁移率最大,被称为快离子,解离度次之的蛋白质则尾随其后,解离度最小的甘氨酸
可溶性蛋白质含量的测定
植物体内可溶性蛋白质含量的测定 植物体内的可溶性蛋白质含量是一个重要的生理生化指标,如在研究每一种酶的作用时常以比活(酶活力单位/毫克蛋白质,unIT/Mg ProTeIn)表示酶活力大小及酶制剂纯度,这就需要测定蛋白质含量。常用的测定方法有LoWry法和考马斯亮蓝G-250染色法,本实验将分别介绍这两种方法。 方法一:LoWry法(劳里法) 【原理】 LoWry法是双缩脲法(BIureT)和福林酚法(FolIn-酚)的结合与发展。其原理是蛋白质溶液用碱性铜溶液处理后,碱性铜试剂与蛋白质中的肽键作用产生双缩脲反应,形成铜—蛋白质的络合盐。再加入酚试剂后,在碱性条件下,这种被作用的蛋白质上的酚类基团极不稳定,很容易还原酚试剂中的磷钨酸和磷钼酸(PHosPHoMolyBdATe &PHosPHoTungsTATe),使之生成磷钨蓝和磷钼蓝的混合物。这种溶液蓝色的深浅与蛋白的含量成正相关,所以可以用于蛋白质含量的测定。LoWry法除使肽链中酪氨酸、色氨酸和半胱氨酸等显色外,还使双缩脲法中肽键的显色效果更强烈,其显色效果比单独使用酚试剂强3~15倍,约是双缩脲法的100倍。由于肽键显色效果增强,从而减少了因蛋白质种类不同引起的偏差。LoWry法适于微量蛋白的测定,对多个样品同时测定较为方便。但对不溶性蛋白和膜结合蛋白必须进行预处理(如加入少量的SDS)。
1.双缩脲法的原理双缩脲(NH2-CO-NH-CO-NH2)在碱性溶液中可与铜离子产生紫红色的络合物,这一反应称为双缩脲反应。因为蛋白质中有多个肽键,也能与铜离子发生双缩脲反应,且颜色深浅与蛋白质的含量的关系在一定范围内符合比尔定律,而与蛋白质的氨基酸组成及分子量无关,所以可用双缩脲法测定蛋白质的含量。 双缩脲反应主要涉及肽键,因此受蛋白质特异性影响较小。且使用试剂价廉易得,操作简便,可测定的范围为1~10Mg蛋白质,适于精度要求不太高的蛋白质含量的测定,能测出的蛋白质含量须在约05Mg以上。双缩脲法的缺点是灵敏度差、所需样品量大。干扰此测定的物质包括在性质上是氨基酸或肽的缓冲液,如TrIs缓冲液,因为它们产生阳性呈色反应,铜离子也容易被还原,有时出现红色沉淀。 2.福林-酚法的原理该方法是双缩脲法的发展,包括两步反应: (1)在碱性条件下,蛋白质与铜作用生成蛋白质—铜络合物。 (2)此络合物将试剂磷钼酸—磷钨酸(FolIn试剂)还原,混合物深蓝色(磷钼蓝和磷钨蓝混合物),颜色深浅与蛋白质含量成正比。此方法操作简便,灵敏度比双缩脲法高100倍,定量范围为5~100μg蛋白质。FolIn试剂显色反应由酪氨酸、色氨酸、半胱氨酸引起,因此样品中若含有酚类、柠檬酸和巯基化合物,均有干扰作用。此方法的缺点是有蛋白质的特异性影响,即不同蛋白质因络氨酸、色氨酸含量的不同而使显色强度稍有不同,标准曲线也不是严格的直线形式。