CLASS-VP 多点(内标法)校正标准曲线制作

CLASS-VP内标法定量操作流程
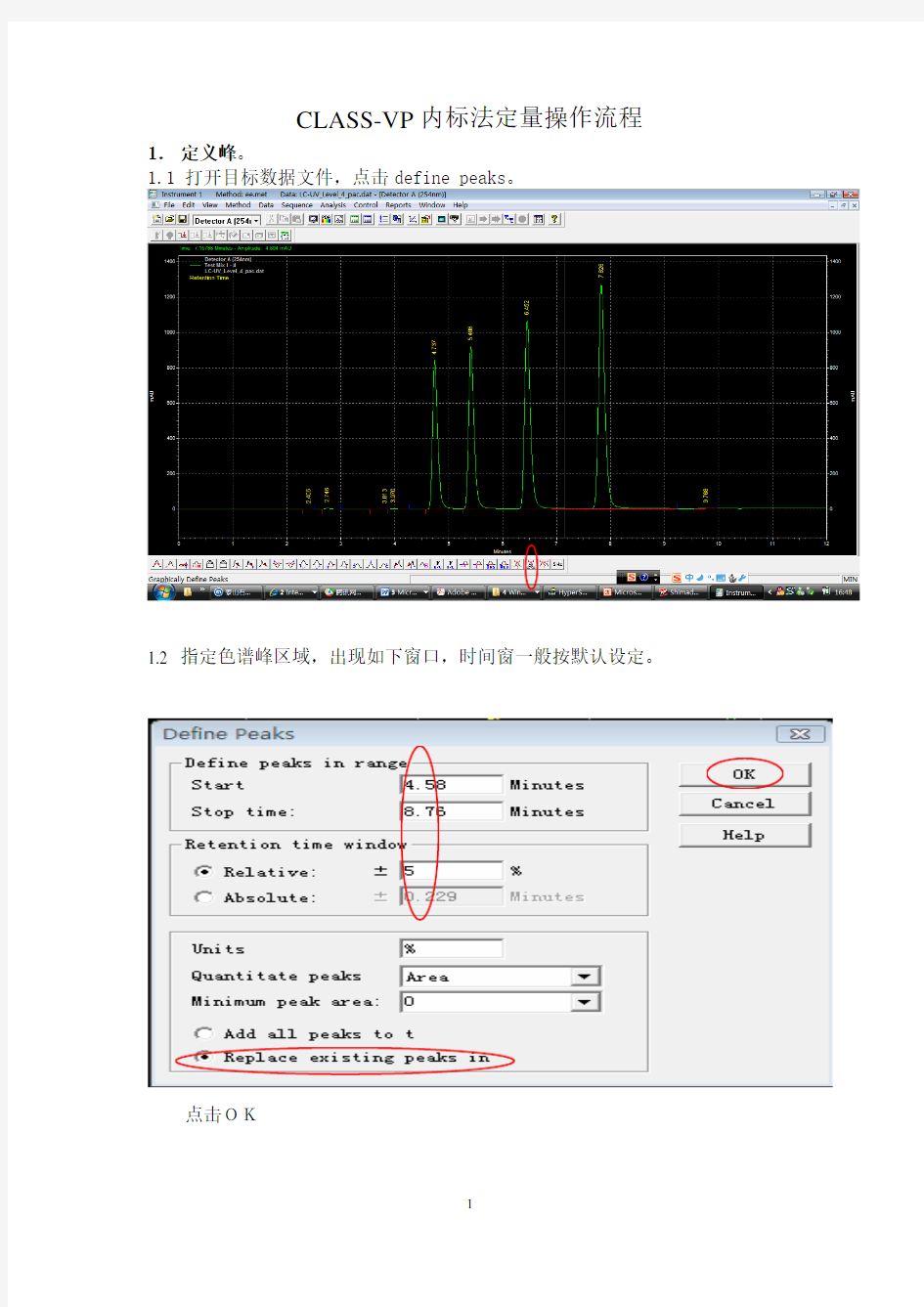
1.定义峰。
1.1 打开目标数据文件,点击define peaks。
1.2 指定色谱峰区域,出现如下窗口,时间窗一般按默认设定。
点击OK
2、编辑峰表(选择Method菜单下 peak/groups)
2.1指定内标法峰:3点校正为例,如2号峰为内标峰,内标峰号ISTD.ID# 如表中设定。选择fit type、输入各组分浓度等。
2.2 方法储存.File/Method/Save As…,命名如IS-test.met,点击保存。
3、建立标准曲线(多点校正时需建立序列表)3.1、建立序列表 File/sequence/new
弹出点击OK
3.2 编辑序列表。调入待校正的标准文件、方法文件(2.2中命名保存的文件)、level值1、2、3等及选择Run Type(第一行选择clear all Calibration和begin calibration,最后一行end calibration)等。
3.3储存序列表:File/Sequence/Save As 序列表文件名为IS-test.seq
点击OK
3.4 运行sequence (sequence/process )
点击Start。
4、查看校准曲线 (Method/Review Calibration 或点击标准曲线工具按钮)
5、计算未知样品浓度
5.1后处理方式
5.1.1打开未知样品数据文件(File/data/open )
点击open
5.1.2查看结果(Reports/View/Internal Standard(Det A))
5.2、实时采集未知样品数据并计算结果
5.2.1未知样品进样 (Control/Single run)
点击Start,进样,采集数据
5.2.2查看结果(reports/view/Internal standard(Det A))
考马斯亮蓝标准曲线制作
标准曲线制作—考马斯亮蓝法测蛋白质含量 一、标准曲线 一般用分光光度法测物质的含量,先要制作标准曲线,然后根据标准曲线查出所测物质的含量。因此,制作标准曲线是生物检测分析的一项基本技术。 二、蛋白质含量测定方法 1、凯氏定氮法 2、双缩脲法 3、Folin-酚试剂法 4、紫外吸收法 5、考马斯亮蓝法 三、考马斯亮蓝法测定蛋白质含量—标准曲线制作 (一)、试剂: 1、考马斯亮蓝试剂: 考马斯亮蓝G—250 100mg溶于50ml 95%乙醇,加入100ml 85% H3PO4,雍蒸馏水稀释至1000ml,滤纸过滤。最终试剂中含0.01%(W/V)考马斯亮蓝G—250,4.7%(W/V)乙醇,8.5%(W/V)H3PO4。 2、标准蛋白质溶液: 纯的牛血清血蛋白,预先经微量凯氏定氮法测定蛋白氮含量,根据其纯度同0.15mol/LNaCl (0.9%NaCl)配制成100ug/ml蛋白溶液。 (二)、器材: 1、722S型分光光度计使用及原理()。 2、移液管使用()。 (三)、标准曲线制作: 1、 2、以A595nm为纵坐标,标准蛋白含量为横坐标(六个点为10ug、20 ug、30 ug、
40 ug、50 ug、60 ug),在坐标轴上绘制标准曲线。 1)、利用标准曲线查出回归方程。 2)、用公式计算回归方程。 3)、或用origin作图,测出回归线性方程。即A595nm=a×X( )+6 一般相关系数应过0.999以上,至少2个9以上。 4)、绘图时近两使点在一条直线上,在直线上的点应该在直线两侧。 (四)、蛋白质含量的测定: 样品即所测蛋白质含量样品(含量应处理在所测范围内),依照操作步骤1操作,测出样品的A595nm,然后利用标准曲线或回归方程求出样品蛋白质含量。 一般被测样品的A595nm值在0.1—0.05之间,所以上述样品如果A595nm值太大,可以稀释后再测A595nm值,然后再计算。 (五)、注意事项: 1、玻璃仪器要洗涤干净。 2、取量要准确。 3、玻璃仪器要干燥,避免温度变化。 4、对照:用被测物质以外的物质作空白对照。
标准曲线的绘制样本
标准曲线绘制 在分析化学实验中, 常见标准曲线法进行定量分析,一般情况下的标准工 作曲线是一条直线。 标准曲线的横坐标(X)表示能够精确测量的变量(如标准溶液的浓度),称为 普通变量,纵坐标(Y)表示仪器的响应值(也称测量值,如吸光度、电极电位 等), 称为随机变量。当X取值为X1, X2,……Xn时,仪器测得的丫值分别为丫1, 丫2,……Yn。将这些测量点Xi, Yi描绘在坐标系中,用直尺绘出一条表示X 与丫之间的直线线性关系,这就是常见的标准曲线法。用作绘制标准曲线的标准物质,它的含量范围应包括试祥中被测物质的含量,标长准曲线不能任意延。用作绘制标准曲线的绘图纸的横坐标和纵坐标的标度以及实验点的大小均不能太 大或太小,应能近似地反映测量的精度。 由于误差不能完全避免,实验点完全落在工作曲线的的情况是极少的,特别是在误差较大时,实验点比较分散,它们一般并不在同一条直线上,这样凭直觉很难判断怎样才能使所连接的直线对于所有实验点来说误差是最小的,当前较好的方法是对实验点(数据)进行回归分析。 研究随机现象中变量之间相关关系的数理统计方法称为回归分析,当自变 量只有一个或X与丫在坐标图上的变化轨迹近似一直线时,称为一元线性回归。 甦2.6.1 —元线性回归方程的求法 确定回归直线的原则是使它与所有测量数据的误差的平方和达到极小值 设回归直线方法为 9 (2 - 15)
式中a表示截距,b表示斜率 9 (2 - 15)
假设Xi和Yi (i=1,2,3, ……,n)是变量X和Y的一组测量数据。对于每一个Xi值,在直线(卩“+^ )上都有一个确定的旳“从X】值。但哲值与X 轴上Xi处的实际测定值Yi是不相等的, 与Yi之差为: 筈厂& +返AY’F-碍(2—佝 上式表示与直线()的偏离程度,即直线的误差程度。如果全部n个测定引起的总偏差用£(节厂印'表示,则偏差平方和s为 (2 - 17) 在所有直线中,偏差平方和s最小的一条直线就是回归直线,即这条直线 的斜率b和截距a应使s值达到最小,这种要使所有数据的偏差平方和达到最小 的求回归直线法称为最小二乘法。 根据数学分析的极值原理,要使s达到最小,对式(2 —17)中的a、b分别 求偏微分后得到 (2 —18) (2 —19) 是所有变量Xi和Yi的平均值。由于计算离均差较麻烦,可将式(2 — 18)变换为 n是测量的次数,也就是坐标图中实验点的数目。 (2 —20)
标准曲线的作法
标准曲线的作法
标准曲线的作法 (1)标准液浓度的选择:在制备标准曲线时,标准液浓度选择一般应能包括待测样品的可能变异最低与最高值,一般可选择5种浓度。浓度差距最好是成 倍增加或等级增加,并应与被测液同样条件下显色测定。 (2)标准液的测定:在比色时,读取光密度至少读2-3次,求其平均值,以 减少仪器不稳定而产生的误差。 (3)标准曲线图的绘制:一般常用的是光密度一浓度标准曲线。 ①用普通方格纸作图。图纸最好是正方形(长:宽=l:1)或长方形(长:宽=3 : 2),以横轴为浓度,纵轴为光密度,一般浓度的全距占用了多少格,光密度的全距也应占用相同的格数。 在适当范围内配制各种不同浓度的标准液,求其光密度,绘制标准曲线, 以浓度位置向上延长,光密度位置向右延长、交点即为此座标标点。然后,将 各座标点和原点联成一条线,若符合Lambert-Beer氏定律,则系通过原点的 直线。 ②若各点不在一直线,则可通过原点,尽可能使直线通过更多点,使不在 直线上的点尽量均匀地分布在直线的两边。 ③标准曲线绘制完毕以后,应在座标纸上注明实验项目的名称,所使用比 色计的型号和仪器编号、滤光片号码或单色光波长以及绘制的日期、室温。 ④绘制标准曲线:一般应作二次或三次以上的平行测定,重复性良好曲线 方可应用。 ⑤绘制好的标准曲线只能供以后在相同条件下操作测定相同物质时使用。 当更换仪器、移动仪器位置、调换试剂及室温有明显改变时,标准曲线需重新 绘制。 ⑥标准曲线横坐标的标度:从标准液的含量换算成待测液的浓度。 1.5 原子吸收光谱分析的定量方法 原子吸收光谱分析是一种动态分析方法,用校准曲线进行定量。常用的定量方法有标准曲线法、标准加入法和浓度直读法。如为多通道仪器,可用内标法定量。在这些方法中,标准曲线法是最基本的定量方法。 1.5.1 标准曲线法 前面已经指出,原子吸收光谱和原子荧光光
Excell软件绘制ELISA标准曲线
怎么用Excell软件绘制ELISA标准曲线 许多试剂检测都涉及到标准曲线的问题,究竟如何绘制或制作标准曲线呢? 用Excell和SPSS的软件能做出来吗?,怎么操作?能一起求出计算公式吗? 有没有专门的软件来处理呢?介绍几种? 希望有这方面经验的介绍自己的经历,与大家分享,“与众同乐才是真的快了” 的确,标准曲线做的好与坏会直接影响到实验的结果,甚至是关系到实验的成败。 首先,做标准曲线样品检测时有几个问题需要注意: 1、样品的浓度等指标是根据标准曲线计算出来的,所以首先要把做标准曲线看作是比做正式实验还要重要的一件事,否则后面的实验结果无从谈起。 2、设置标准曲线样品的标准浓度范围要有一个比较大的跨度,并且要能涵盖你所要检测实验样品的浓度,即样品的浓度要在标准曲线浓度范围之内,包括上限和下限。而对于呈S型的标准曲线,尽量要使实验样品的浓度在中间坡度最陡段,即曲线几乎成直线的范围内。 3、最好采用倍比稀释法配制标准曲线中的标准样品浓度,这样就能够保证标准样品的浓度不会出现较大的偏离。 4、检测标准样品时,应按浓度递增顺序进行,以减少高浓度对低浓度的影响,提高准确性。 5、标准曲线的样品数一般为7个点,但至少要保证有5个点。 6、做出的标准曲线相关系数因实验要求不同而有所变动,但一般来说,相关系数R至少要大于0.98,对于有些实验,至少要0.99甚至是0.999.
怎样绘制标准曲线? 标准曲线浓度得到后,可通过计算器、Excell或SPSS统计软件进行绘制,并得到相关的回归方程(即计算公式和相关系数(回归系数,个人认为,Excell 软件比较好用一些;SPSS也行,不过是英文的,初学者不是很容易掌握;计算器嘛太麻烦了,所以现在一般不用。 双抗夹心法ELISA拟和曲线: 拟和曲线: 打开EXCEL软件;在工作表中 输入第一行:浓度值,如0 10 50 100 400 输入第二行:该浓度下的调整后的od值,如0 0.586 1.397 1.997 3.42 选择这些输入的数据,用插入里的图表按钮,进入图表向导,在“标准类型”中选择“xy散点图”;在“子图表类型”中选择“折线散点图”,按“下一步”;选择“系列产生在行”,按“下一步”;数据标志,可以填写:如数据y 轴,OD值;数据x轴,浓度;按下一步,点击完成。可得曲线图。 单击曲线,按右键,选择“添加趋势线”,在类型中,选择多项式;在选项中,选择显示公式,选择显示R平方值。得到公式和R平方值。 也可以用上面说的方法,在公司已经提供的图表上,双击图表,把它输入到图表的数据中,就可以拟和新的曲线。 计算浓度: 举例:第一次实验: 标准曲线为:
标准曲线数据表
5.含量的测量 【数据记录】 ● 称量信息: 10片片重W=4.390g W 平均=439 mg 片粉重W ’=0.073g A 样品=0.126 ● 计算: 带入公式:A=0.0059X+0.006, 得X (浓度)=20.34 ug/ml 百分含量%=C*V*n/(片粉重量)=20.34*25*100/10^6*0.073=69.65% ● 分析: 若将最后一点带入曲线,R=0.997,线形不如只带前五点好。因此,省去第六点,这样线形在三个9,线形良好 【结果与结论】该批次阿司匹林片剂中原料药的百分含量为69.65%。 6.溶出度与溶出速率 【数据记录】 乙酰水杨酸片的溶出速率测定数据 取样时间(min) 吸光 度 实测浓度(ug/ml) 校正浓度(ug/ml) 溶出百分率 F(%) X Y 5 0.103 16.6 16.6 0.243737051 1.609438 -1.27523 10 0.18 32 32.083 0.471073241 2.302585 -0.45113 15 0.244 44.8 45.043 0.661364336 2.70805 0.079578 25 0.339 63.8 64.267 0.943629461 3.218876 1.056334 35 0.38 72 72.786 1.068713553 3.555348 #NUM! 45 0.418 79.6 80.746 1.185589874 3.806662 #NUM! (1) F=50%, 带入线形公式计算得, t=10.04 min , Lnt=2.31——T50 (2) F=63.2%,带入线形公式计算得, t=12.94 min , Lnt=2.56——Td (3)m 值计算:过M 点(1,0)的直线为, y=1.426x-1.426, 所以,m=-1.426 (4)溶出度测定: F(45min)= 118.6% > 70%,溶出百分率复合药典规定 【结果】 T50为2.31,时间10.04min , Td 为2.56,时间为12.94min, m=-1.426, F(45min)为118.6%>70% 【结论】该片剂的溶出度符合药典规定,溶出速率符合Welbull 方程,溶出较快,较均匀。 标准曲线数据表 序号 浓度(ug/ml) 吸光度 1 10 0.066 2 20 0.123 3 30 0.183 4 40 0.242 5 50 0.301 6 100 0.551
标准曲线的制作方法
标准样品的准备 由于你做的是基因表达分析,样品的准备就比较简单了,因为你要知道都是相对的数值(你的对照样品和实验样品中基因表达的比值),这样就不需要知道精确的拷贝数,所以标准品也就无须知道精确的拷贝数,只需知道稀释的倍数就可以了。 你实验中的标准品可以是来源比较丰富的细胞或组织的RNA转录得到的cDNA。将这种cDNA进行梯度的稀释,可以是稀释10倍,100,1000,10000等倍(具体到你的实验要根据具体的情况来调整稀释倍数。最好能作一个预实验来看看什么样的稀释倍数比较适合你的这种基因的扩增)。而对于各个稀释倍数我们要对它的拷贝数进行赋值,这个值当然不是标准品中真实含有的基因数量(在基因表达分析中也不需要),而是我们根据稀释倍数给每一个稀释度人为赋予的拷贝数,这只是为了方便实验最终结果的计算而已。比如我们把前面稀释十倍的样品赋值为10000个拷贝,100倍的赋值为1000个拷贝依次类推把10000倍的赋为10等。要注意,赋值的数目的倍数差异和你稀释的倍数是一样的,比如前面是10稀释,后面赋值也是10倍变化。 如何做标准曲线 在定量实验中标准品是要和你的未知样品一起进行定量实验的,这样在实验结束,无论是标准品还是未知样品都将跑出曲线,获得Ct值。那么我们先可以把未知样品放到一边,对于标准品来说,我们既获得了Ct值,还知道他们的拷贝数(虽然这个拷贝数是我们自己赋予的)。这样我们可以通过标准品的Ct值和拷贝数做一条标准曲线(以拷贝数为横坐标,而Ct值为纵坐标)。一旦作出了标准曲线,而未知样品的Ct值我们知道(通过实验求得的),这时候就在标准曲线上进行定位,就可以得到未知样品的拷贝数了。 事实上,操作起来没有那么复杂,你只需要告诉软件你的哪个孔是标准品,哪个是未知样品,以及标准品的拷贝数等必要信息,软件会自动帮你把标准曲线和未知样品的拷贝数计算出来。 举例来说如何进行设置 先进入软件,在板设置中选择所放样品的位置,并标上unknow或standard等信息。 选择要使用的荧光染料。
标准曲线法与标准加入法的区别
1 标准曲线法 1.1标准曲线法的计算公式 在一定条件下,标准曲线是一条直线,直线的斜率和截距可以用最小二乘法求得。现在好多仪器软件都能自动生成标准曲线,所以一小部分版友不清楚标准曲线的具体计算方法。本人查找了一些资料,找到标准曲线的斜率和截距的计算方法,和大家分享。 工作曲线可以用一元线性方程表示: y=a+bx (1) 式中,x为标准溶液的浓度,y为相应的吸光度。 使用最小二乘法确定的直线称为回归线,a,b称为回归系数。 b 为直线的斜率,可由下式求得:
1.2 灵敏度 灵敏度是指该方法对单位浓度或单位量的待测物质的变化所引起的响应量变化的过程。一般用标准曲线的斜率b为方法的灵敏度,b越大,灵敏度越高。 不要小看灵敏度,用处可大了。灵敏度由于仪器的不同,实验条件等的不同,在不断的变化。但在一定的实验条件下,灵敏度相对还是比较稳定的。所以,建议对灵敏度也做个质量控制图,具体做法见我另一个帖子。 每次标准曲线做好以后,观察灵敏度是否在一定的范围内维持稳定。如果发现灵敏度突然降低,就需要考虑,是否是仪器出问题了。火焰首先考虑雾化器是否堵塞,石墨炉首先考虑石墨管是否是烧坏。解决方法是用通丝清理雾化器,更换石墨管后继续测定标准曲线,带灵敏度稳定后进行样品测定。还有,在打开一瓶新的标准溶液的时候,在仪器进行维修以后,一定要注意灵敏度的变化。 1.3线性范围 线性范围这个大家都比较清楚,主要从相关系数r看,一般要求r大于等于三个九。我在这里和大家分享的是,我想了好久才想开的一个问题。之前看好多书上
后来专门做了好多次实验,还是一直是直线在,只是有时候浓度高时,线性不好,高浓度点不在标准曲线上,而是在标准曲线的下面,而且离拟合的标准曲线比较远。遇到这种情况,标准曲线的线性相关系数就很差,有时候才一个九,如图2所示。最后我终于想明白了,如果自己用手动拟合的话,用平滑的曲线去连接所有点的话,你就会发现,如果在线性范围内,连接起来就是直线,如果超出了线性范围,连接起来就是一条弯曲的曲线。从弯曲的拐点开始,就已经超出了线性范围,如图3所示。 所以,判断是否超出线性范围,个人觉得不是看是否弯曲,软件用最小二乘法拟合的一次曲线,永远是直线。要看你的高浓度点是否在拟合的标准曲线上或离得比较近,相关系数是否在三个九以内。如果不在,从拐点开始的那个点,就超出了线性范围,应该删除,从新拟合。 1.4检出限 检出限是指对某一特定的分析方法在给定的置信水平内可以从样品中检测待测物质的最小浓度或最小量。
应用EXCEL绘制ELISA标准曲线及计算样本浓度
应用EXCEL绘制ELISA标准曲线及计算样本浓度 整体思路:在ELISA实验结束后,我们得到的是经酶标仪读出的标准品以及样本的OD值。其中,标准品的浓度已知,我们可以利用标准品的OD值及其相对应的浓度,以OD值为横坐标,浓度为纵坐标,做一条标准曲线,并用公式表示出来。这样,我们把样本的OD值以X值代入,便可求出Y值,即样本浓度。下面我们就一步步来实现这个目标: 1. 首先,将数据整理好输入Excel,分别为经酶标仪读出标准品的OD值及其对应的浓度值。 2. 拖动鼠标选取完成的数据区,并点击图表向导,在图表类型中选“XY散点图”,并选择子 图表类型的“散点图”(第一个没有连线的)。如下图所示。
3. 点击“下一步”,出现如下图界面。如是输入是如本例横向列表的就不用更改,如 果是纵向列表就改选“列”。
在做ELISA标准曲线,并通过此曲线求样本浓度时,因样本OD值是已知的,因此,我们需要将OD值设为X。根据上图我们可以看到,横坐标X值为OD值,纵坐标Y为标准品浓度,这正是我们想要的。 4.有时需要我们手动选择X值,这时可以点击“系列”来更改,如下图。
如果要对横坐标和纵坐标进行掉换,我们可以就点击X值和Y值文本框右边的小图标,结果如下图:
出现上图后,拖动鼠标选取正确的数据区域以调整X或Y。 5.调好之后,然后点击“下一步”出现图表选项界面,如下图。 6.点击“下一步”,在弹出的窗口再点击“完成”现在一张带标准值的完整散点图就已经完成,如下图。
7.到了这一步,散点图便完成了。我们可能会问,曲线在哪里啊? 其实很简单,先点击图上的标准值点(记住一定要点选上),然后单击右键,点击“添加趋势线”。如下图:
运用Ecel做标准曲线
E x c e l绘制标准曲线全图片教程 随着计算机的日益普及,越来越多的检验工作者希望能从一些烦琐的工作中解脱出来,如:绘制标准曲线、绘制质控图、计算检测值等等。当然借助检验科办公系统理论上是最方便的,但很多单位是没有检验科办公系统的。其实借助Microsoft的Excel电子表格工具对检验工作也会带来很大的便利。 Excel是Microsoft offices系统的重要组成,它是界于WORD字处理软件与ACCESS数据库软件之间的电子表格工具,功能十分强大,特别适合于日常工作使用。使用得好,完全比目前所有的检验科办公系统优秀。 现就先介绍一下如何使用Excel绘制标准曲线。 首先,将数据整理好输入Excel,并选取完成的数据区,并点击图表向导,如下图所示。 点击图表向导后会运行图表向导如下图,先在图表类型中选“XY散点图”,并选了图表类型的“散点图”(第一个没有连线的)。 点击“下一步”,出现如下图界面。如是输入是如本例横向列表的就不用更改,如果是纵向列表就改选“列”。 如果发现图不理想,就要仔细察看是否数据区选择有问题,如果有误,可以点击“系列”来更改,如下图。 如果是X值错了就点击它文本框右边的小图标,结果如下图:
出现上图后,如图在表上选取正确的数据区域。然后点击“下一步”出现图表选项界面,如下图,上应调整选项,以满足自己想要的效果。 点击“下一步”,现在一张带标准值的完整散点图就已经完成,如下图。 完成了散点图,现在需要根据数据进行回归分析,计算回归方程,绘制出标准曲线。其实这很简单,先点击图上的标准值点,然后按右键,点击“添加趋势线”。如下图。 由于本例是线性关系,在类型中选“线性”如下图 点击“确定”,标准曲线就回归并画好了。 标准曲线是画好了,可是我们怎么知道回归后的方程是什么样呢这了简单,点击趋势线(也就是我们说的标准曲线)然后按右键,选趋势线格式,如下图:在显示公式和显示R平方值(直线相关系数)前点一下,勾上。再点确定。好了,现在公式和相关系数都出来了。如图:呵R的平方达,线性相当好。 可是有时候有的项目是成指数增加的,散点图如下图, 将左下方的对数刻度选中,确定。完整的一个半对数标准曲线就做好了。 利用Excel制作标准曲线简单吧如果认真调整参数可以得到不同的效果,大家多研究一下吧。 有人说标准曲线是拿来用的,要是能输入信号值就可以得出浓度那就好了,其实使用Excel也能很自如的处理。具体请听下回分解。
HPLC标准曲线的制作
HPLC标准曲线的制作 你可以随便弄一个浓度进一针样品看一看你这个样品的吸收度如何,再根据你样品的吸收度配置适当的底浓度样品逐个稀释这样可以连检测线定量限一起做了,一举3得(最后将你得到的峰面积根据你的进样浓度做一个线性回归就行啦,线性好的话R的平方一般接近于1 。 请问下各位在上样的时候是进同浓度的不同体积的样液绘制标准曲线好还 是先配好不同浓度的样液再以相同体积进样好呢, 这2个有什么区别吗, 请你看看分析化学书,有关精密度和线性的关系就知道了,实在不行,可以查看2005版药典一部附录新药质量标准的技术要求项下。自己看看就知道了。 我觉得进不同浓度相同体积好.因为你进样体积不同.在同样的方法下,系统的 各项参数可能会发生变化.而且你要通过进样体积的变化来控制浓度范围,这样也不大可行.一般我们进样的体积是5到20微升.而这个狭小的范围我们能调控的浓度线性范围非常窄.而通过事先调配不同浓度的话,就简单可行,而且浓度范围可以任意去控制.在实际操作中,老师也一直是教我们通过控制不同浓度来制作标准曲线的.以上只是个人的粗浅看法,欢迎高手们前来批评指正!!!!!!!!!! 前面几个站友说得都很好。我简单再补充一点自己的看法。 一般而言,还是不同浓度进相同体积做标曲是最规范的做法,而且可以有效避免人为误差。比如说,你的一个浓度配错了,如果以这个浓度为基准进不同的体积,会导致你最后的结果会整体偏大或偏小。所以要特别注意。 但实际工作中,如果你们的产品做得比较成熟了,而且做实验的经验比较丰富,大家为了省事还是多采用配一个浓度进不同的体积。 以进样量做标准曲线和以不同浓度进样相同体积做标准曲线差别不大。
标准曲线制作考马斯亮蓝法测蛋白质含量
标准曲线制作考马斯亮蓝法测蛋白质含量
标准曲线制作—考马斯亮蓝法测蛋白质含量 一、标准曲线 一般用分光光度法测物质的含量,先要制作标准曲线,然后根据标准曲线查出所测物质的含量。因此,制作标准曲线是生物检测分析的一项基本技术。 二、蛋白质含量测定方法 1、凯氏定氮法 2、双缩脲法 3、Folin-酚试剂法 4、紫外吸收法 5、考马斯亮蓝法 三、考马斯亮蓝法测定蛋白质含量—标准曲线制作 (一)、试剂: 1、考马斯亮蓝试剂: 考马斯亮蓝G—250 100mg溶于50ml 95%乙醇,加入100ml 85% H3PO4,雍蒸馏水稀释至1000ml,滤纸过滤。最终试剂中含0.01%(W/V)考马斯亮蓝G—250,4.7%(W/V)乙醇,8.5%(W/V)H3PO4。 2、标准蛋白质溶液: 纯的牛血清血蛋白,预先经微量凯氏定氮法测定蛋白氮含量,根据其纯度同0.15mol/LNaCl配制成100ug/ml蛋白溶液。 (二)、器材: 1、722S型分光光度计使用及原理()。 2、移液管使用()。 (三)、标准曲线制作: 1、 试管编号0 1 2 3 4 5 6 100ug/ml标准蛋白(ml)0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.15mol/L NaCl (ml) 1 0.9 0.8 0.7 0.6 0.5 0.4 考马斯亮蓝试剂(ml) 5 5 5 5 5 5 5 摇匀,1h内以1号管为空白对照,在595nm处比色 A595nm 2、以A595nm为纵坐标,标准蛋白含量为横坐标(六个点为10ug、20 ug、30 ug、
药品的配制(磷酸缓冲液的配制) 一、药品的配制步骤 (一)、实验准备: 1、准备所需的药品和玻璃仪器。 2、洗涤。(怎样洗涤算干净?) (二)、计算: 1、百分比浓度计算: 1)、G/V比 例如配1% NaCl,称1g NaCl溶于100ml 水。 2)、V/V比: 例如配75%乙醇100ml,75%×100%=100%×X, X=75ml。取75ml无水乙醇,加25ml蒸馏水。 乙醇:乙醚:丙酮=2:1:2配500ml,各取200 ml,100 ml,200 ml混合。3)G/V比:用的较少,如计算灰分中某种元素如Fe的含量。 2、摩尔浓度计算:注:药品的分子量一般在标签中注明。 1)、0.1M或0.1mol/L NaCl配100ml。 M=质量/体积(L)称取NaCl0.1×0.1×40=0.4g 摩尔数=G(g)/摩尔质量2)、0.1mMNaCl配100ml mM=毫摩尔数/体积(L)称取NaCl0.1×0.1×40=0.4g 毫摩尔数=G(mg)/摩尔质量 3)、0.1uNaCl配100ml mM=微摩尔数/体积(L)称取NaCl0.1×0.1×40=0.4mg 微摩尔数=G(ug)/摩尔质量称取NaCl0.1×0.1×40=0.4ug 3、混合溶液配制的计算: 如配3uMEDTA,2.25mM NBT以及60uM 溶液100ml,用50mM磷酸缓冲液配制。 注意:1、分别标定体积计算 2、分别配制再混合,但总体积不能为100ml
分析化学中的标准曲线
分析化学中的标准曲线
在分析化学实验中,常用标准曲线法进行定量分析,通常情况下的标准工作曲线就是一条直线。 标准曲线的横坐标(X)表示可以精确测量的变量(如标准溶液的浓度),称为普通变量,纵坐标(Y)表示仪器 的响应值(也称测量值,如吸光度、电极电位等),称为随机变量。当X取值为X1, X2,…… Xn时,仪器测得的Y值分别为Y1, Y2, …… Yn。将这些测量点Xi, Yi描绘在坐标系中,用直尺绘出一条表示X与Y之间的直线线性关系,这就就是常用的标准曲线法。用作绘制标准曲线的标准物质,它的含量范围应包括试祥中被测物质的含量,标准曲线不能任意延长。用作绘制标准曲线的绘图纸的横坐标与纵坐标的标度以及实验点的大小均不能太大或太小,应能近似地反映测量的精度。 由于误差不能完全避免,实验点完全落在工作曲线的的情况就是极少的,尤其就是在误差较大时,实验点比较分散,它们通常并不在同一条直线上,这样凭直觉很难判断怎样才能使所连接的直线对于所有实验点来 说误差就是最小的,目前较好的方法就是对实验点(数据)进行回归分析。 研究随机现象中变量之间相关关系的数理统计方法称为回归分析,当自变量只有一个或X与Y在坐标图上的变化轨迹近似一直线时,称为一元线性回归。 2、6、1一元线性回归方程的求法 确定回归直线的原则就是使它与所有测量数据的误差的平方与达到极小值,设回归直线方法为 (2-15) 式中a表示截距,b表示斜率。 假设Xi与Yi (i=1,2,3,……,n)就是变量X与Y的一组测量数据。对于每一个Xi值,在直线( )上都有一个确定的值。但值与X轴上Xi处的实际测定值Yi就是不相等的,与Yi之差为: (2-16) 上式表示与直线()的偏离程度,即直线的误差程度。如果全部n个测定引起的总偏差用 表示,则偏差平方与s为 (2-17) 在所有直线中,偏差平方与s最小的一条直线就就是回归直线,即这条直线的斜率b与截距a应使s值达到最小,这种要使所有数据的偏差平方与达到最小的求回归直线法称为最小二乘法。 根据数学分析的极值原理,要使s达到最小,对式(2-17)中的a、b分别求偏微分后得到
标准曲线的绘制吸光度标准曲线绘制
标准曲线的绘制-吸光度标准曲线绘 制 生物化学实验报告ALT与其吸光度的标准曲线绘制 采集样本:广西医科大学口腔医学2016级13班四个组中7组生物化学实验数据采集时间:2016年11月15日2016~2016上学期第十一周周一下午采集人:何洁梅 一、几组ALT与其吸光度的标准曲线数据记录 ALT活力单位A520吴修团1组A520黎丁菱1组A520杨璇璇1组A520谢晓兰2组A520莫雪玲2组A520李文良3组A520文全海4组 00000000
二、各采集样本汇总图 样本1测定得待测血清ALT活力单位为50U/L 样本2测定得待测血清ALT活力单位为 97U/L 样本3测定得待测血清ALT活力单位为 135U/L 样本4测定得待测血清ALT活力单位为 70U/L 样本5测定得待测血清ALT活力单位为 148U/L 样本6测定得待测血清ALT活力单位为
45U/L 样本7测定得待测血清ALT活力单位为 98U/L 四、采集数据处理结果分析 1.数据总结 样本编号测定的ALT活力单位是否大于40U/L正常/非正常 150是非正常 297是非正常 3135是非正常 470是非正常 5148是非正常 645是非正常 798是非正常 平均值92均为“是”均为“非正常” 2.针对数据处理结果的分析 采集的7组数据经标准曲线测量后,得到的ALT活力单位值均大于40,即均为非正常值,综上,认为待测血清中ALT 含量超于正常值。 3.针对源数据的分析
采集的7组数据中样本4、5、6的数据经画图后可基本分布呈过原点的线性关系,符合理论规律,但其他的数据误差较大。另外,比较符合理想标准曲线的4、5、6样本的三个ALT活力单位值也存在较大的出入。 4.经分析,总结可能的误差来源如下 配置丙酮酸标准溶液、底物溶液、磷酸缓冲液的混合溶液时,丙酮酸标准溶液的剂量都很小,容易造成误差。 加入2,4—二硝基苯肼的时间可能有误差,保温的时间,以及加入NaOH 以停止反应的时间都有可能有偏差,容易造成较大。如何用EXCEL绘制标准曲线 Excel是Microsoft offices系统的重要组成,它是界于WORD字处理软件与ACCESS数据库软件之间的电子表格工具,功能十分强大,特别适合于日常工作使用。使用得好,完全比目前所有的检验科办公系统优秀。 现就先介绍一下如何使用Excel绘
ELISA标准曲线制作方法.pdf
ELISA标准曲线制作方法 一般而言,我们拟合ELISA标准曲线选用比较经典的Curve Expert 1.3或者Curve Expert 1.4软件。现在我们以Curve Expert 1.4为例,对ELISA标准曲线绘制方法进行详述。 Generally speaking, the standard fitting curve of ELISA is based on the classic software of Curve Expert 1.3 or Curve Expert 1.4. Now we will use the Curve Expert 1.4 to illustrate how to construct the ELISA standard curve. 1.点击Curve Expert 1.4,打开应用程序,截图界面如下: Click Curve Expert 1.4, and then open the application programs. You will see the following screen shot.
2.在X轴输入标准品的OD值,Y轴输入相应的标准品的浓度,截图界面如下: Input the value of OD on the X-axis against the concentration of samples on the Y-axis. The following screen shot will appear like this.
3.单击上图界面中的图标,出现如下界面 Click the icon showed on the above screen shot and you will see the following picture. 4.单击上图界面中的ALL OFF 按钮,出现如下界面 Click the ‘All Off’ button , you can see the following screen shot.
标准曲线不确定度的分析精选文档
标准曲线不确定度的分 析精选文档 TTMS system office room 【TTMS16H-TTMS2A-TTMS8Q8-
标准曲线不确定度的分析 曹涛 (深圳市通量检测科技有限公司,广东深圳 518102) 摘要:本文阐述了标准曲线不确定度分析的通用方法,讲解了标准曲线不确定度的分析步骤和方法。并且通过检测过程中经典的几个项目的标准曲线的不确定度实例分析,来论证影响不确定度分析的因素;通过实验数据证明:参与标准曲线的点的个数和样品测定的次数越多,标准不确定度越小;并且样品测定的结果越靠近标准曲线的重点,标准不确定度越小;标准曲线的线性越好,斜率越高,标准不确定度越好; 关键词:标准曲线,不确定度 中图分类号:文献标识码:A文章编号: 作者简介:曹涛(1987-06-04),男,汉族,陕西省汉中市,中山大学本科毕业,研究食品中营养成分、添加剂、农兽药残留的检测分析;E-mail Analysis of Standard Curve’s Uncertainty Cao Tao (Shenzhen Total-Test Technology Co., Ltd, Shenzhen 518102)
Abstract: This paper expounds a general method of the analysis of standard curve’s uncertainty and explains the steps of the analysis of standard curve’s uncertainty. Prove the effect of the factors of uncertainty analysis through the testing process in classical several items of standard curves of uncertainty analysis. Experiments demonstrate: Improving the te st’s times of the standard curve and sample can make the standard’s uncertainty small. And The results of sample are more close to the center of the standard curve, the standard’s uncertainty is smaller. T he standard curve is linear in the better, and the slope is higher, the standard uncertainty can be smaller. Key word: the standard curve. uncertainty 前言:仪器分析中线性回归标准曲线测定方法,利用被测物质相应的信号强度与其浓度成正比关系,通过测定已知浓度的溶液(即标准溶液)的信号强度,通过 最小二乘法将响应值和浓度的对应的线性关系拟合成一条直线,再根据未知样品的响应值推算出对应的浓度;然而测得的所有点未必全部都落在标准曲线上(除非线性相关系数r=1),因此得到的标准曲线本身具备相应的不确定性,而通过标准曲线去计算得到的浓度值就不可避免的具备不确定性,而且这个不确定性往往是整个实验不确定度的最大来源;因此对标准曲线计算不确定度非常有必要; 1 标准曲线不确定度分析的概念和计算 标准曲线的不确定度忽略标准溶液的不确定度的引入
标准曲线的作法
标准曲线的作法 (1)标准液浓度的选择:在制备标准曲线时,标准液浓度选择一般应能包括待测样品的可能变异最低与最高值,一般可选择5种浓度。浓度差距最好是成倍增加或等级增加,并应与被测液同样条件下显色测定。 (2)标准液的测定:在比色时,读取光密度至少读2-3次,求其平均值,以减少仪器不稳定而产生的误差。 (3)标准曲线图的绘制:一般常用的是光密度一浓度标准曲线。 ①用普通方格纸作图。图纸最好是正方形(长:宽=l:1)或长方形(长:宽=3 : 2),以横轴为浓度,纵轴为光密度,一般浓度的全距占用了多少格,光密度的全距也应占用相同的格数。 在适当范围内配制各种不同浓度的标准液,求其光密度,绘制标准曲线,以浓度位置向上延长,光密度位置向右延长、交点即为此座标标点。然后,将各座标点和原点联成一条线,若符合Lambert-Beer氏定律,则系通过原点的直线。 ②若各点不在一直线,则可通过原点,尽可能使直线通过更多点,使不在直线上的点尽量均匀地分布在直线的两边。 ③标准曲线绘制完毕以后,应在座标纸上注明实验项目的名称,所使用比色计的型号和仪器编号、滤光片号码或单色光波长以及绘制的日期、室温。 ④绘制标准曲线:一般应作二次或三次以上的平行测定,重复性良好曲线方可应用。 ⑤绘制好的标准曲线只能供以后在相同条件下操作测定相同物质时使用。当更换仪器、移动仪器位置、调换试剂及室温有明显改变时,标准曲线需重新绘制。 ⑥标准曲线横坐标的标度:从标准液的含量换算成待测液的浓度。 1.5 原子吸收光谱分析的定量方法 原子吸收光谱分析是一种动态分析方法,用校准曲线进行定量。常用的定量方法有标准曲线法、标准加入法和浓度直读法。如为多通道仪器,可用内标法定量。在这些方法中,标准曲线法是最基本的定量方法。 1.5.1 标准曲线法 前面已经指出,原子吸收光谱和原子荧光光谱分析是一种相对测定方法,不能由分析信号的大小直接获得被测元素的含量,需通过一个关系式将分析信号与被测元素的含量关联起来。校正曲线就是用来分析信号(即吸光度)转换为被测元素的含量(或浓度)的“转换器”,此转换过程成为校正。之所以要进行校正,是因为同一元素含量在不同的试验条件下所得到的分析信号是不同的。校准曲线的制作方法是,用标准物质配制标准系列溶液,在标准条件下,测定各标准样品的吸光度值Ai,以吸光度值Ai(i=1,2,3,4,5)对被测元素含量ci(i=1,2,3,4,5)绘制校准曲线A=f(c)。在同样条件下,测定样品的吸光度值Ax,根据被测元素的吸光度值Ax从校准曲线求得其含量Ci。校准曲线如图1-4所示。 校准曲线的质量直接影响校准效果和样品测定结果的准确度。正确地制作一条高质量校准曲线是非常重要的,为此需要:(1)合理的设计校准曲线;(2)分析信号的准确测定;(3)正确绘制校准曲线。 首先,从数理统计的观点出发合理设计校准曲线。根据一组实验点绘制校准曲线所遵循的原则是最小二乘原理,即要让实验点随机地分布在校正曲线的周围,并有尽可能多的实验点落在标准曲线上,使得由这些实验点绘制的标准曲线的标准偏差最小。从校准曲线的置信范围考虑,当实验点数目,<4时,置信系数较大且变化速率较快,置信范围较宽,由校正
标准曲线的绘制
标准曲线绘制 在分析化学实验中,常用标准曲线法进行定量分析,通常情况下的标准工作曲线是一条直线。 标准曲线的横坐标(X)表示可以精确测量的变量(如标准溶液的浓度),称为普通变量,纵坐标(Y)表示仪器的响应值(也称测量值,如吸光度、电极电位等),称为随机变量。当X取值为X1, X2,…… Xn时,仪器测得的Y值分别为Y1, Y2, …… Yn。将这些测量点Xi, Yi描绘在坐标系中,用直尺绘出一条表示X与Y 之间的直线线性关系,这就是常用的标准曲线法。用作绘制标准曲线的标准物质,它的含量范围应包括试祥中被测物质的含量,标准曲线不能任意延长。用作绘制标准曲线的绘图纸的横坐标和纵坐标的标度以及实验点的大小均不能太大或太小,应能近似地反映测量的精度。 由于误差不能完全避免,实验点完全落在工作曲线的的情况是极少的,尤其是在误差较大时,实验点比较分散,它们通常并不在同一条直线上,这样凭直觉很难判断怎样才能使所连接的直线对于所有实验点来说误差是最小的,目前较好的方法是对实验点(数据)进行回归分析。 研究随机现象中变量之间相关关系的数理统计方法称为回归分析,当自变量只有一个或X与Y在坐标图上的变化轨迹近似一直线时,称为一元线性回归。 2.6.1一元线性回归方程的求法 确定回归直线的原则是使它与所有测量数据的误差的平方和达到极小值,设回归直线方法为 (2-15) 式中a表示截距,b表示斜率。 假设Xi和Yi (i=1,2,3,……,n)是变量X和Y的一组测量数据。对于每一个Xi值,在直线() 上都有一个确定的值。但值与X轴上Xi处的实际测定值Yi是不相等的,与Yi之差 为: (2-16) 上式表示与直线()的偏离程度,即直线的误差程度。如果全部n个测定引起的总偏 差用表示,则偏差平方和s为 (2-17)
标准曲线制作
1 葡萄糖测定 (1)1mg/mL 葡萄糖标准液 小烧杯取分析纯葡萄糖置于烘干箱中烘干至,于分析天平中准确称取80℃烘至恒重的分析纯 葡萄糖10mg,置于小烧杯中,加少量蒸馏水溶解后,转移到10mL 容量瓶中,用蒸馏水定 容至10mL,混匀,4℃冰箱中保存备用。 (2)3,5-二硝基水杨酸(DNS)试剂 将6.3g DNS (分析天平中称取于小烧杯)和262mL 2M NaOH 溶液(大量筒称取250ml+ 小量筒称取12ml),加到500mL(量杯量取)含有185g 酒石酸钾钠的热水溶液中(在微波 炉中加热1min溶解),再加5g 结晶酚和5g 亚硫酸钠,搅拌溶解,冷却后加蒸馏水定容至1000mL,贮于棕色瓶中备用。 1.制作葡萄糖标准曲线 取7 支10mL 具塞刻度试管编号,按表1分别加入浓度为1mg/mL 的葡萄糖标准液、蒸馏水 和3,5-二硝基水杨酸(DNS)试剂,配成不同葡萄糖含量的反应液。 1mg/ml葡萄糖标准溶液蒸馏水 DNS 葡萄糖含量光密度值 管号(ml)(ml)(ml)(mg/ml)(OD540) 0 0 1 0.75 0 1 0.1 0.9 0.75 0.01 2 0.2 0.8 0.75 0.02 3 0.3 0.7 0.75 0.03 4 0.4 0.6 0.7 5 0.04 5 0.5 0.5 0.75 0.05 6 0.6 0.4 0.75 0.06 将各管摇匀,取试管架置于水浴锅中,在沸水浴中准确加热5min,取出,冷却至室温,用蒸馏水定容至10mL,加塞后颠倒混匀,在分光光度计上进行比色(分光光度计预热20分钟)。调波长540nm,用0 号管调零点,测出1~6号管的光密度值。以光密度值为纵坐标,葡萄糖含量(mg)为横坐标,在坐标纸上绘出标准曲线。 发酵取样:1ml 样品+0.75mlDNS 沸水浴5min,冷却后,定容到10ml. 540nm测定 2 酒精度测定 称取优级纯无水乙醇0.1000g (取小烧杯置于分析天平中,去皮,以微量移液枪小心添 加至0.1000g)于100ml 容量瓶中(蒸馏水洗涤小烧杯导入容量瓶), 加水至刻度。此溶液 每毫升相当于1.0mg 乙醇。 5 %重铬酸钾溶液: 称取5g 重铬酸钾(AR) 溶于50ml 水中, 加10ml 浓硫酸(用20ml量 筒量取,边加边用玻璃棒搅拌), 放冷, 取100ml容量瓶,加水至100ml。 试剂均为分析纯, 水为蒸馏水。 分别取乙醇标准溶液(1mg/mL)0, 0.2,0.4,0.6, 0.8, 1.0mL于带刻度的10mL试管中,分别加入重铬酸钾1.0ml,用蒸馏水补足至5.0ml,在100 ℃水浴中加热10min (加塞防止乙 醇挥发), 取出用流水冷却5min ,充分反应,测吸光度。