纳米金负载型催化剂的研究进展

2004年6月 贵 金 属 Jun. 2004 第25卷第2期 Precious Metals V ol. 25, No. 2
收稿日期:2003-09-08 基金项目:云南省自然科学基金项目资助(No..2000E 0008Z, 2002E0080M ) 作者简介:周 华,男,现正在攻读博士学位。 ﹡联系人:董守安,男,研究员。
纳米金负载型催化剂的研究进展
周 华,董守安*(昆明贵金属研究所,云南 昆明 650221)
Advances in the Research of Supported Gold Particle Catalysts
ZHOU Hua, DONG Shouan (Kunming Institute of Precious Metals, Kunming, Yunnan 650221, China )
Abstract: Nanotechnology has transformed gold from a chemical inert metal into a new and effective
catalyst with potential industrial application prospect. In the present paper, a dvances in the recent
research of supported gold particle catalyst ware reviewed in detail, including catalyst preparation,
catalytic performance and typical applications as well as reaction mechanism, in order to understand
further unique properties of gold catalyst.
Keywords: Catalysis chemistry ;Supported gold catalyst ;Preparation method ;Catalytic performance ;
Applications ;Reaction mechanism
摘 要: 纳米技术已经把化学惰性的金变成新的、具有工业应用前景的催化剂。作者从纳
米金催化剂的制备、催化性能、典型应用以及催化机理等方面对近4年来金催化剂的研究现
状进行了较全面的综评。以便进一步加深对金催化剂的认识。
关键词:催化化学;负载金催化剂;制备方法;催化性能;应用;反应机理 中图分类号:TG146.3 文献标识码:A 文章编号:1004-0676(2004)02-0048-09
长期以来,人们一直认为金是一种化学惰性的金属, 因为块状金几乎不产生任何化学吸附而无
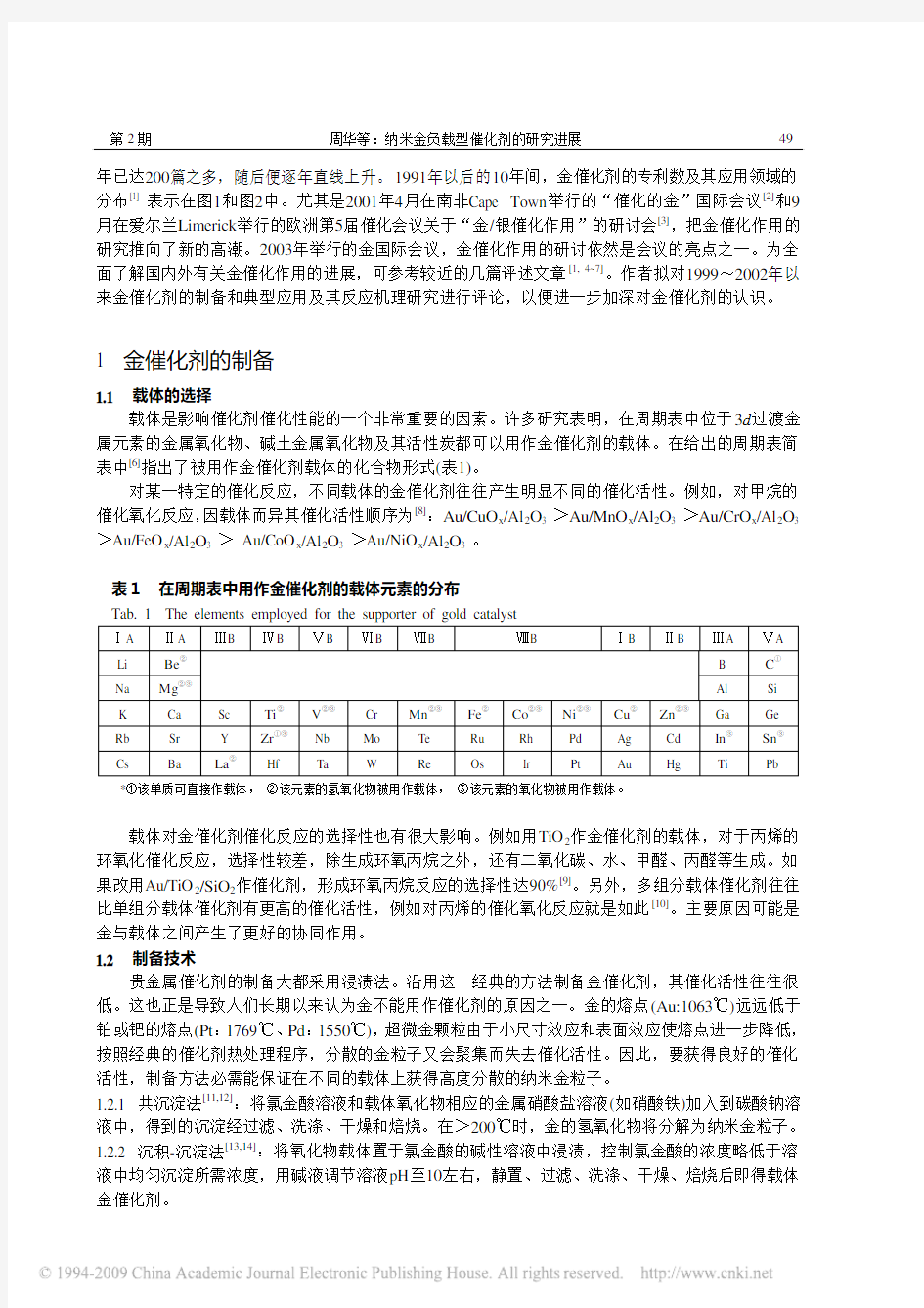
催化作用。然而,纳米技术已经彻底地改变了人们对金催化作用的认识 ,当把它高度分散于过渡金属氧化物载体上而形成超微粒子时,化学惰性的金变成了高活性的催化剂。自20世纪90年代至今,金催化作用的研究取得了飞速的发展,工程应用上也很受重视。关于金催化研究所发表的论文在1998
图 1 金催化专利数目图 Fig. 1 Patent numbers of catalysys based on gold
图 2 金催化专利(1991~2001)分布领域图
Fig. 2 Distribution of gold catalysis patents in various area
第2期 周华等:纳米金负载型催化剂的研究进展 49
年已达200篇之多,随后便逐年直线上升。1991年以后的10年间,金催化剂的专利数及其应用领域的分布[1] 表示在图1和图2中。尤其是2001年4月在南非Cape Town举行的“催化的金”国际会议[2]和9月在爱尔兰Limerick举行的欧洲第5届催化会议关于“金/银催化作用”的研讨会[3],把金催化作用的研究推向了新的高潮。2003年举行的金国际会议,金催化作用的研讨依然是会议的亮点之一。为全面了解国内外有关金催化作用的进展,可参考较近的几篇评述文章[1, 4~7]。作者拟对1999~2002年以来金催化剂的制备和典型应用及其反应机理研究进行评论,以便进一步加深对金催化剂的认识。
1 金催化剂的制备
1.1 载体的选择
载体是影响催化剂催化性能的一个非常重要的因素。许多研究表明,在周期表中位于3d过渡金属元素的金属氧化物、碱土金属氧化物及其活性炭都可以用作金催化剂的载体。在给出的周期表简表中[6]指出了被用作金催化剂载体的化合物形式(表1)。
对某一特定的催化反应,不同载体的金催化剂往往产生明显不同的催化活性。例如,对甲烷的催化氧化反应,因载体而异其催化活性顺序为[8]: Au/CuO x/Al2O3 >Au/MnO x/Al2O3 >Au/CrO x/Al2O3 >Au/FeO x/Al2O3 >Au/CoO x/Al2O3 >Au/NiO x/Al2O3 。
表1 在周期表中用作金催化剂的载体元素的分布
Tab. 1 The elements employed for the supporter of gold catalyst
ⅠA ⅡA ⅢB ⅣB ⅤB ⅥB ⅦB ⅧB ⅠB ⅡB ⅢA ⅤA Li Be② B C①Na Mg②③Al Si K Ca Sc Ti②V②③Cr Mn②③Fe②Co②③Ni②③Cu②Zn②③Ga Ge Rb Sr Y Zr①③Nb Mo Te Ru Rh Pd Ag Cd In③Sn③Cs Ba La②Hf Ta W Re Os Ir Pt Au Hg Ti Pb *①该单质可直接作载体,②该元素的氢氧化物被用作载体,③该元素的氧化物被用作载体。
载体对金催化剂催化反应的选择性也有很大影响。例如用TiO2作金催化剂的载体,对于丙烯的环氧化催化反应,选择性较差,除生成环氧丙烷之外,还有二氧化碳、水、甲醛、丙醛等生成。如果改用Au/TiO2/SiO2作催化剂,形成环氧丙烷反应的选择性达90%[9]。另外,多组分载体催化剂往往比单组分载体催化剂有更高的催化活性,例如对丙烯的催化氧化反应就是如此[10]。主要原因可能是金与载体之间产生了更好的协同作用。
1.2 制备技术
贵金属催化剂的制备大都采用浸渍法。沿用这一经典的方法制备金催化剂,其催化活性往往很低。这也正是导致人们长期以来认为金不能用作催化剂的原因之一。金的熔点(Au:1063℃)远远低于铂或钯的熔点(Pt:1769℃、Pd:1550℃),超微金颗粒由于小尺寸效应和表面效应使熔点进一步降低,按照经典的催化剂热处理程序,分散的金粒子又会聚集而失去催化活性。因此,要获得良好的催化活性,制备方法必需能保证在不同的载体上获得高度分散的纳米金粒子。
1.2.1 共沉淀法[11,12]:将氯金酸溶液和载体氧化物相应的金属硝酸盐溶液(如硝酸铁)加入到碳酸钠溶液中,得到的沉淀经过滤、洗涤、干燥和焙烧。在>200℃时,金的氢氧化物将分解为纳米金粒子。
1.2.2 沉积-沉淀法[13,14]:将氧化物载体置于氯金酸的碱性溶液中浸渍,控制氯金酸的浓度略低于溶液中均匀沉淀所需浓度,用碱液调节溶液pH至10左右,静置、过滤、洗涤、干燥、焙烧后即得载体金催化剂。
50 贵 金 属 第25卷
1.2.3 胶体负载法[15~18]:在PVP 或PV A 等高分子保护下,用水合肼或甲醛等试剂还原HAuCl 4溶液制得金溶胶。然后负载于氧化物或活性炭载体上,经干燥、焙烧后得金催化剂。
1.2.4 离子交换法[19,20]:将HAuCl 4的水溶液与HY 分子筛共热,使之与分子筛作用以取代载体表面或内部的H ,再经焙烧、还原等活化处理后可以制得Y 型分子筛作载体的金催化剂。
1.2.5 化学气相沉积法[21~23]:在真空条件下,将挥发性的金属有机化合物通过蒸汽导入并使之吸附于比表面积较高的金属氧化物载体上,然后于473K 下在空气中焙烧,使有机金属化合物分解为小颗粒的金。这种方法可以广泛地应用于各种不同的金属氧化物载体上,制得的催化剂活性一般都比较高。
除了上述几种方法之外,用于金催化剂制备的还有金属有机配合物固载法[24~27]、共溅射法[28]、光化学沉积法[29]、合金氧化法[30,31] 和溶剂化金属原子浸渍法[32]等。
2 负载金催化剂的催化性能和应用
金负载在氧化物载体上所形成的一系列新的金催化剂,能应用于各式各样的反应体系。从图2中可以看出,目前金催化剂的主要应用集中在下述3大领域。
2.1 环境工程
为了控制水和空气质量,使用在环境温度下工作的非均相催化剂是最富有吸引力的途径。在大气环境中,消除汽车排放废气中的氮氧化物和CO 是很紧迫的事情,在这方面有许多使用金催化剂富有成效的研究。载金的过渡金属氧化物催化剂体系作为低温三效催化剂已经在汽车尾气排放控制方面表现出潜力,因为它能同时降低碳氢化合物和CO 的起活温度。金催化剂在低温即具有催化CO 氧化的高催化活性,且能在潮湿的空气中工作,它能将CO 从空气中除去。据悉, 新型的载金催化剂有望在今后5年内商业化。
2.1.1 一氧化碳的氧化:CO 的氧化是负载型金催化剂研究和应用的一个经典反应,文献比较丰富[33 ]。在温度<400K ,金催化剂的催化活性比其它贵金属催化剂均高。如Au/Fe 2O 3催化剂在-70℃仍能进行CO 的氧化反应[28]。适用该反应的金催化剂可选用的载体非常多,包括TiO 2、Fe 2O 3、MO X 等[34~37],但是载体不同,催化剂的活性相差甚远。当CO 的催化转化率为95%时,金颗粒的尺寸、载体和反应温度之间的相互关系如图3所示[10]。制备方法也是十分重要的。用沉积-沉淀法制备的金催化剂的催化活性要远大于用浸渍法和光化学沉积法制备的。在制备过程中加入助剂也有利于催化
剂性能的改善。
2.1.2 氮氧化物的还原:在汽车排放的废气中,NO
和烃类的含量一般都比较高,严重威胁人类健康和破坏自然环境。而载金催化剂对于氮氧化物的还原反应,具有很高的催化活性[38~40]。以Fe 2O 3为载体的金催化剂可将反应温度降至50℃左右。用NiFe 2O 4、CoFe 2O 4担载的金催化剂,活性较Fe 2O 3为载体要高许多,甚至能够在室温下进行反应。这也反映了载体金属氧化物对N-O 、C-O 键的活化能力。
(1) 2NO+2CO=N 2+CO 2: 对于该反应,金催化剂的活性和选择性明显高于其它贵金属催化剂。在50℃以下消耗差不多计量的CO ,N 2是主要的还原产物 [41]。但O 2的出现不利于反应的进行,因为O 2比NO 更容易和CO 反应。
(2) 用烃类还原NO :该反应的历程为:NO →NO 2→N 2 。当烃类为丙烷、丙烯、乙烯及乙烷时,
图3 在不同载体上CO 转化率为95%所需温 度与金粒径的关系
Fig. 3 Temperature needed for 95% CO conversion vs gold particle size on different supports
(▲ Au/Al 2O 3、300℃, ■ Au/Al 2O 3、600℃, ◆ Au/Al 2O 3 、900℃, ● Au/MOx/Au/Al 2O 3
)
第2期 周华等:纳米金负载型催化剂的研究进展 51 金催化剂对此类反应具有较高的催化活性。而且CO 和H 2的存在有利于NO 的还原。
由于从机动车辆排出的尾气温度,尤其在起活温度时低于150℃[39,41],因而金催化剂的低温催化活性引起了人们的关注。催化剂的商业应用,必须考虑潮湿气氛和氧的影响,而金催化剂能在潮湿的条件下获得更好的催化效果。
2.1.3 烃类燃烧:汽车发动机刚启动时排出大量烃类物质,金催化剂可以加速烃类物质的燃烧[42~43]。
对于该类反应,金颗粒越小,催化活性越高。采用共沉淀法得到的5%Au/Co 3O 4,显示了很高的催化氧化甲烷的能力。然而,随着反应的进行,催化剂上的金粒子逐渐被烧结而失活。总体上看,对于饱和烃类的催化燃烧,金催化剂的活性与铂、钯催化剂相当;对于不饱和烃类的燃烧,金催化剂的活性不如铂、钯催化剂。Au/Fe 2O 3对三甲基胺的催化燃烧反应有很高的催化活性,此外,该催化剂能够用于消除大气和厕所中的异味[41]。
2.1.4 有机卤化合物污染的消除:有机卤化合物(如CCl 2F 2)是破坏大气臭氧层的罪魁祸首。对于CCl 2F 2和CH 3Cl 化合物的氧化分解反应,Au/Co 3O 4或Au/Al 2O 3催化剂的活性与Pt 催化剂相当[7]。Pd-Au/C 催化剂的选择性较高,可以达到86%。影响金催化剂对此类反应的催化活性主要在于制备方法 [42]。
2.2 化学工程和精细化工领域
非均相金催化剂除了对大气污染的净化有重要作用之外,对C1化学催化反应、不同类型的加氢反应、水煤气转化反应和液相氧化反应都具有催化作用,是很有工业应用前景的催化剂。
2.2.1 低温水煤气转换反应:水煤气转换反应是廉价获得氢气的一个重要的化工过程。已有2种商业化催化剂:Fe 2O 3/Cr 2O 3和CuO/ZnO/Al 2O 3,使用温度分别是563~723K和483~513K。同这2种催化剂相比,金催化剂明显的优势在于它的低温(73K)
催化活性[5,43,44]。
影响该反应催化剂活性的因素比较多[45],主要
有:①制备方法的影响。不同的制备方法,即使是相同的载体,催化剂的活性也相差甚大,如表2所示[37]。因此选择制备方法应充分考虑载体的性质,目的是提高金与载体之间的相互作用。②金粒子尺寸和分散性的影响。金颗粒的最佳尺寸为3~5nm 。采用相同的制备方法,用不同载体的催化剂,如Au/Ce 2O 3、Au/Co 3O 4和Au/TiO 2会产生不同的金粒子分散度和催化效果。 2.2.2 丙烯的环氧化:环氧丙烷是一种重要的化工原料,全世界每年需求量在3500kt 左右,市场价值70亿美元。现有生产环氧丙烷的工艺复杂,且要使用氯气或有机过氧化物,造成环境污染。在H2、O2存在下,负载在TiO 2上的金催化剂具有催化丙烯环氧化的能力,因此引起了学术界和
化工界极大关注[46~49]。
Au/TiO 2/SiO 2催化剂可以将丙烯一步氧化生成环氧丙烷,在473K下转化率接近5%,选择性很高,水是主要副产物。影响催化剂活性的因素主要有 [5]:①催化剂的制备方法, 浸渍法制备的催化剂基本上不能实现丙烯的环氧化,反应只生成H 2O 和CO 2。然而,用DP 法制备的催化剂则可以使目标产物的选择性>90%。原因可能是DP 法使金颗粒与载体TiO 2产生强烈的相互作用。②载体的影响,Au/TiO 2催化转化率仅约2%, 使用复合氧化物TiO 2/SiO 2作为载体,转化率提高了一倍多。③
表2 金催化剂的制备方法、载体和金粒子尺寸对水煤气转换反应的影响
Tab. 2 Effect of average size of gold particle of the
gold-bearing catalysts based on different supports prepared by different method on water-gas shift reaction 催化剂 制备方法* 金颗粒尺寸(nm) t 1/2(℃) Au/Fe 2O 3 CP 4.0 160 Au/Fe 2O 3 CP >1.0 280 Au/Fe 2O 3 MCP 3.5 150 Au/Co 3O 4 CP 8.0 235 Au/Co 3O 4 MCP 7.0 200 Au/TiO 2 DP <5.0 130 Au/TiO 2 MDP <3.0 300
Au/ZrO 2 DP <5.0 150 Au/ZrO 2 MDP <3.0 300 Au/CeO 2 DP 4.5 200 *CP 为共沉淀;DP 为沉积沉淀;MCP 和MDP 为修饰的前2种方法
52 贵 金 属 第25卷
粒子尺寸的影响,金颗粒<2nm ,反应产物不是环氧丙烷而是丙烷。④添加剂的影响,实验证明,Na + 和Cl - 的存在对反应具有促进作用。该反应用于工业过程,还有待进一步提高反应转化率的研究。 2.2.3 邻二醇类的选择性液相氧化:非均相金催化剂常用于气-固相的催化氧化反应。最近发现负载于活性炭上的金催化剂在邻二醇类的液相选择性氧化方面,特别是用于制备羟基乙酸、乳酸和葡萄糖酸显示很高的催化活性[50,51], 转化率>90%,而且选择性很好。尤其对葡萄糖的氧化反应,在低温低压条件下,转化率≈100%,且具有很好的抗失活能力。另外,金负载于Al 2O 3或TiO 2上时对该反应也显示出很好的催化活性。值得指出的是,如图4所示,金负载于Al 2O 3上时,金颗粒尺寸越小活性越高;而负载于活性炭上时,金颗粒为7~8nm 时催化活性最佳。 同其它贵金属催化剂相比,Au/C 催化剂具有如下
优点:①不易引起C—C键的断裂,而易选择性催化一个羟基基团的氧化;② 反应的转化率高,选择性好;③具有较好的抗失活能力。例如,经过4次反应循环,5%Pt/C和5%Pd/C催化剂的选择性分别由79%、82%降至48%、26%,而Au/C催化剂仍>90%。
2.2.4 二氧化碳的氢化:甲醇是一种重要的基本化工原料。随着对温室效应的关注,人们对于利用CO 2氢化合成甲醇产生了浓厚的兴趣。对于CO 和CO 2的氢化反应,Au/TiO 2、Au/Fe 2O 3和Au/ZnFe 2O 4在150-400K 具有催化活性,其中CO 2比CO 更容易氢化而制得甲醇[6]。特别是Au/ZnO 和Au/ZnFe 2O 4催化剂对该反应有较高的选择性,其效果可以和商业上所使用的铜系列催化剂相媲美。而对于CO 的氢化,只有Au/ZnO 催化剂才能达到和铜系列催化剂相类似的催化效果。但如果反应温度过高,得到的产物不是甲醇而是甲烷[52]。
2.2.5 烃类的选择性氢化:总的来说,烃类的加氢反
应对催化剂的结构不是很敏感的。作为金催化剂的载
体,例如Al 2O 3、SiO 2、TiO 2等对该反应的催化活性影
响不大。金催化剂显著的特点是对部分氢化具有非常
好的选择性[53,55],例如由丁二亚基化合物制丁烯衍生
物、乙炔制乙烯等。对α,β不饱和醛的氢化反应,
只要催化剂上金纳米颗粒>2nm ,那它对C=O 基团的部分氢化的选择性达到40%~50%。对于乙炔加氢反应,金颗粒为3nm 时,催化剂的活性最高,如图5所示[5]。 2.3 燃料电池 新型燃料电池驱动的机车能耗低,无有害气体产生。其中有一类新型燃料电池是通过催化剂的催化作
用把液态有机烃类转化成氢气,后者在燃烧过程中与氧作用生成水,但美中不足的是此类燃料电池装置仍很笨重,在商业化前仍然有许多技术问题有待解决。采用新型金催化剂有望能减轻装置的重量、缩小体积,并在可接受的技术性能指标下,有助于降低燃料电池制造成本。研究结果显示,金催化剂不仅使装置能在较低的温度下工作,而且还可从氢气流中除掉微量的CO, 从而提高燃料电池的效率。在水煤气转化反应中,可以为燃料电池提供氢源。
图4 在Au/Al 2O 3和Au/C 催化剂上Au 粒子尺寸对邻二醇氧化的影响 Fig. 4 Dependence of catalytic activity of Au/Al 2O 3 and Au/C catalysts on the diameter of Au particles for the oxidation of glycols (a 氧化物载体, b 炭载体) 图5 Au 粒子尺寸对乙炔加氢反应的影响 Fig. 5 Dependence of catalytic activity of catalyst on the diameter of Au particle for
acetylene hydrogenation
第2期 周华等:纳米金负载型催化剂的研究进展 53
3金催化剂的催化机理
块状金对于绝大多数分子来说是惰性的,然而纳米技术的引入,粒子表面的化学吸附及反应活性随结构明显地发生变化。这首先应归结于纳米结构的3大效应[56],即小尺寸效应、表面效应和量子尺寸效应。其中前2种效应的影响是最为突出的。由于表面原子周围缺乏相邻的原子,使得颗粒出现大量的剩余悬键而具有不饱和的性质,即配位数不足,表面活性位增多,和迅速增加的表面能一起构成起催化作用的关键因素。再就是作者曾在‘纳米技术中的金元素’一文[57]中简要介绍的金的相对论的收缩效应。金的6S电子具有很高的稳定性,增加了范德华作用力和金表面的物理吸附能,也导致了纳米金粒子异常的催化性能。
3.1 CO的化学吸附
CO吸附在金颗粒表面是CO氧化反应最重要的一步,有许多学者对此进行了深入的探讨。用沉积沉淀法制得的金催化剂有3个位置可以对CO产生吸附,即CO可以吸附于单质金粒子表面、金的氧化物表面和金原子簇之上[5]。Haruta[58]等研究了CO在Au/TiO2上的吸附情况,结果表明:在-20~50℃内有90%的CO在Au/TiO2上的等温吸附符合Langmuir方程;不可逆吸附的CO约占10%,恰好与在吸附过程中金颗粒表面上产生的CO2数量相吻合。在不同的吸附位置和温度范围内,反应具有不同活化能,分温度段讨论的反应机理如下列反应所示[5]:
200K以下于金粒子表面上的反应:
Au(s)-CO + Auδ+(s)-O2δ- CO2(g)+Au(s)+Auδ+(s)-Oδ-;Au(s)-CO + Auδ+(s)-Oδ- CO2(g)+2Au(s) ;Au(p)-CO + Auδ+(p)-O2δ- CO2(g)+Au(p)+Auδ+(p)-Oδ-;Au(p)-CO + Auδ+(p)-Oδ- CO2(g)+2Au(p) 在300K以上于金粒子周边界面间的反应:
Au(p)-CO + TiO2(p)-O2δ- CO2(g)+TiO2(p)+ e-+Auδ+(p)-Oδ-;Au(p)-CO + Auδ+(p)-Oδ- CO2(g)+2Au(p) 在300K以下所伴随的反应:
Au(p)-CO + TiO2(p)-O2δ-+ e- Au(p)+TiO2(p)-CO32-
其中,(s):台阶(step) (p):边界(perimeter) (g):气相(gas phase)
3.2 O2 的化学吸附
块状金表面不吸附O2 。然而当用各种方式来激活或分解O2分子时,则会产生原子O的吸附。但氧气究竟以何种形式吸附于金催化剂之上,一直是学术界争论的焦点。有学者认为氧气以分子形式吸附于金催化剂表面[59,60],而另一部分学者则认为氧气应以原子形式被吸附[61]。另一争论是O2吸附的位置。Huruta认为O2不可逆吸附于金和载体的界面处。Baiker则认为O2不可逆吸附在金属氧化物的空缺位置上[7]。
4 结语
通过上述讨论可知,金催化剂具有优异的低温催化活性、良好的抗水性、稳定性和湿度增强效应。可望在环保、精细化工和燃料电池方面得到非常广泛的应用。加强金催化剂的研究与开发,无论在学术上还是工业上,都有着重要的意义。国外的研究和应用已经相当广泛和深入,加强金催化剂的研发是我们面临的一项刻不容缓的任务。
参考文献
[1] Corti C W, Holliday R J, Thompson D T. Developing new industrial applications for gold : gold nanotechnology [J].
Gold Bulletin, 2002,35:111-117.
[2] Thompson D T. The international conference on catalytic gold[J]. Gold Bulletin, 2001,34:56-66.
54 贵金属第25卷
[3] Thompson D T. Highlights of the symposium on gold /silver catalysis[J].Gold Bulletin, 2001,34:134-140.
[4] Bond G C. Gold: a relatively new catalyst[J]. Gold Bulletin, 2001,34: 117-119; Catalysis Today, 2002,72: 5-9.
[5] Haruta M, Date M. Advances in the catalysis of Au nanoparticles[J]. Appl.Catal.A,General, 2001, 427-437.
[6] 王东辉, 郝郑平, 程代云等. 负载型金催化剂在化工中的应用[J].化工进展, 2002,21: 462-465.
[7] 齐世学, 邹旭华, 安立敦. 负载型金催化剂[J].化学通报, 2002, 11: 734-741.
[8] Grisel R J H, Nieuwenhuys B E. A comparative study of the oxidation of CO and CH4 over Au/MO x/Al2O3
catalysts[J]. Catalysis Today, 2001, 64: 69-81.
[9] Mul G, Wijnenburg A Z, Linden van der B, et al. Stability and selectivity of Au/TiO2 and Au/TiO2/SiO2 catalysts
in propene epoxidation: an in Situ FT-IR study[J]. Journal of Catalysis, 2001, 201:128-137.
[10] Grisel R, Westrte K-J, Gluhoi A, et al. Catalysis by gold nanoparticles[J]. Gold Bulletin, 2002, 35:39-45.
[11] Golunski S, Rajaram R, Hodge N, et al. Low-temperature redox activity in co-precipitated catalysts: a comparison
between gold and platinum-group metals[J]. Catalysis Today, 2002, 72: 107-113.
[12] Hodge N A, Kiely C J, Whyman R, et al. Microstructural comparison of calcined and uncalcined gold/iron-oxide
catalysts for low-temperature CO oxidation[J]. Catalysis Today, 2002, 72:133-144.
[13] Park E D, Lee J S.Effects of pretreatment conditions on CO oxidation over supported Au catalysts[J]. Journal of
Catalysis, 1999, 186:1-11.
[14] Wolf A, Schüth F. A systematic study of the synthesis conditions for the preparation of highly active gold catalysts
[J]. Appl. Catal. A: General, 2002, 226:1-13.
[15] Seker E, Gulari E. Single step sol-gel made gold on alumina catalyst for selective reduction of NO x under
oxidizing conditions: effect of gold precursor and reaction conditions[J]. Appl.Catal. A: General, 2002, 232: 203-217.
[16] Schimpf S, Lucas M, Mohr C, et al. Supported gold nanoparticles: in-depth catalyst characterization and
application in hydrogenation and oxidation reactions[J].Catalysis Today, 2002, 72: 63-78.
[17] Grunwaldt J-D,Kiener C, W?gerbauer C,et al. Preparation of supported gold catalysts for low-temperature CO
oxidation via "size-controlled" gold colloids[J]. Journal of Catalysis, 1999, 181: 223-232.
[18] Porta F, Prati L, Rossi, M, et al. Metal sols as a useful tool for heterogeneous gold catalyst preparation:
reinvestigation of a liquid phase oxidation[J]. Catalysis Today, 2000, 61:165-172.
[19] Uphade B S,Akita T, Nakamura T, et al. Vapor-phase epoxidation of propene using H2 and O2 over
Au/Ti-MCM-48[J]. Journal of Catalysis, 2002, 209: 331-340.
[20] Riahi G, Guillemot D, Polisset-Thfoin M, et al. Preparation, characterization and catalytic activity of gold-based
nanoparticles on HY zeolites[J]. Catalysis Today, 2002, 72: 115-121.
[21] Choudhary T V, Sivadinarayana C,Chusuei C C, et al. CO oxidation on supported nano-Au catalysts synthesized
from a [Au6(PPh3)6](BF4)2 complex[J]. Journal of Catalysis, 2002,207:247-255.
[22] Kozlova A P,Sugiyama S,Kozlov A I, et al. Iron-oxide supported gold catalysts derived from gold-phosphine
complex Au(PPh3)(NO3): state and structure of the support[J]. Journal of Catalysis, 1998,176: 426-438.
[23] Chen Y J, Yeh C, Deposition of highly dispersed gold on alumina support[J]. Journal of Catalysis, 2001,200:59-68.
[24] 邹旭华,齐世学,贺红军等. 以Au(PPh3) (NO3) 为前体的Au/NiO催化剂的制备及其对CO的催化氧化[J].分
子催化, 2000, 14: 171-174.
[25] 邹旭华,齐世学,安立敦.不同方法制备的负载型金催化剂对CO的催化性能[J]. 中国学术期刊文摘,2000, 6:
505-506.
[26] Kozlova A P,Kozlov A I, Sugiyama S,et al. Study of gold species in iron-oxide-supported gold catalysts derived
from gold-phosphine complex Au(PPh3)(NO3) and As-precipitated wet Fe(OH)3 [J]. Journal of Catalysis, 1999,181:37-48.
第2期 周华等:纳米金负载型催化剂的研究进展 55
[27] Liu H, Kozlov A I,Kozlova A P,et al. Active oxygen species and mechanism for low-temperature CO oxidation
reaction on a TiO2-supported Au catalyst prepared from Au(PPh3)(NO3) and As-precipitated titanium hydroxide [J]. Journal of Catalysis, 1999,185:252-264.
[28] Uematsu T, Fan L, Maruyama T, et al. New application of spray reaction technique to the preparation of supported
gold catalysts for environmental catalysis [J]. Journal of Molecular Catalysis, A, Chemical, 2002, 182-183: 209-214.
[29] Cunningham D A H,V ogel W, Sanchez R M T, et al. Structural analysis of Au/TiO2catalysts by Debye function
analysis [J]. Journal of Catalysis, 1999, 183:24-31.
[30] Espinosa G, Del Angel G, Barbier J, et al. Catalytic behavior and active sites structure of PtAu/Al2O3 bimetallic
catalysts prepared by surface redox reactions [J].Journal of Molecular Catalysis, A, Chemical, 2000, 164: 3-262.
[31] Chandler B D,Schabel A B,Blanford C F, et al. Preparation and characterization of supported bimetallic Pt-Au
particle catalysts from molecular cluster and chloride salt precursors [J]. Journal of Catalysis.1999, 187:367-384.
[32] 吴世华,黄唯平,张守民等. 溶剂化金属原子浸渍法制备高分散Au/TiO2低温CO氧化催化剂[J].催化学报,
2000,21, 419-422.
[33] 王东辉, 程代云, 郝郑平等. 纳米金催化剂上CO低(常)温氧化的研究[J]. 化学进展, 2002, 14: 360-367.
[34] Daté M,Haruta M. Moisture effect on CO oxidation over Au/TiO2catalyst [J]. Journal of Catalysis,
2001,201:221-224.
[35] Lin C-H, Hsu S-H, Lee M-Y,et al.Active morphology of Au/γ-Al2O3-A model by EXAFS [J]. Journal of
Catalysis, 2002, 209: 62-68.
[36] Grisel R J H, Weststrate C J, Goossens A, et al. Oxidation of CO over Au/MO x/Al2O3 multi-component catalysts
in a hydrogen-rich environment [J]. Catalysis Today, 2002, 72:123-132.
[37] Bethke G K, Kung H H. Selective CO oxidation in a hydrogen-rich stream over Au/-Al2O3 catalysts [J]. Applied
Catalysis A, General, 2000, 194-195:43-53.
[38] Ruth K, Hayes M, Burch R, et al. The effects of SO2 on the oxidation of CO and propane on supported Pt and Au
catalysts [J]. Applied Catalysis B, Environmental, 2000,24: 133-138.
[39] Krawczyk K, Motek M. Combined plasma-catalytic processing of nitrous oxide [J]. Applied Catalysis B,
Environmental, 2001, 30: 233-245.
[40] Craenenbroeck J V , Andreeva D, Tabakova T, et al. Spectroscopic analysis of Au-V-based catalysts and their
activity in the catalytic removal of diesel soot particulates [J]. Journal of Catalysis, 2002, 209: 515-527.
[41] Ueda A, Haruta M. Nitric oxide reduction with hydrogen,carbon monoxide,and hydrocarbons over gold catalysts
[J]. Gold Bull., 1999, 32: 3-11.
[42] Bonarowska M, Burda B, Juszczyk W, et al. Hydrodechlorination of CCl2F2 (CFC-12) over Pd-Au/C catalysts [J].
Applied Catalysis B, Environmental, 2001, 35:13-20.
[43] Andreeva D, Idakiev V, Tabakova T, et al. Low-temperature water-gas shift reaction over Au/CeO2 catalysts [J].
Catalysis Today, 2002, 72:51-57.
[44] Tabakova T, Idakiev V, Andreeva D, et al. Influence of the microscopic properties of the support on the catalytic
activity of Au/ZnO, Au/ZrO2, Au/Fe2O3, Au/Fe2O3-ZnO, Au/Fe2O3-ZrO2 catalysts for the WGS reaction [J].
Applied Catalysis A, General, 2000, 202: 91-97.
[45] Andreeva D. Low temperature water gas shift overgold catalysts [J]. Gold Bull., 2002, 35: 82-88.
[46] Mul G,Zwijnenburg A, Van der Linden B, et al. Stability and selectivity of Au/TiO2 and Au/TiO2/SiO2 catalysts
in propene epoxidation: an in situ FT-IR study [J]. Journal of Catalysis, 2001, 201: 128-137.
[47] Uphade B S, Akita T,Nakamura T,et al. Vapor-phase epoxidation of propene using H2 and O2 over
Au/Ti-MCM-48 [J]. Journal of Catalysis, 2002, 209: 331-340.
[48] Stangland E E, Stavens K B, Andres R P,et al.Characterization of gold-titania catalysts via oxidation of propylene to
56 贵金属第25卷
propyleneoxide [J]. Journal of Catalysis, 2000, 191: 332-347.
[49] Uphade B S, Okumura M, Tsubota S, et al. Effect of physical mixing of CsCl with Au/Ti-MCM-41 on the
gas-phase epoxidation of propene using H2 and O2: drastic depression of H2 consumption [J]. Applied Catalysis A, General, 2000, 190: 43-50.
[50] Porta F, Prati L, Rossi M, et al. Metal sols as a useful tool for heterogeneous gold catalyst preparation:
reinvestigation of a liquid phase oxidation [J]. Catalysis Today, 2000, 61: 165-172.
[51] Biella S,Prati L,Rossi M. Selective oxidation of D-glucose on gold catalyst [J]. Journal of Catalysis, 2002, 206:
242-247.
[52] Sakurai H, Haruta M. Carbon dioxide and carbon monoxide hydrogenation over gold supported on titanium, iron,
and zinc oxides [J]. Applied Catalysis A, General, 1995, 127: 93-105.
[53] Hutchings G J. Gold catalysis in chemical processing [J]. Catalysis Today, 2002, 72: 11-17.
[54] Sárkány A, Horváth A, Beck A. Hydrogenation of acetylene over low loaded Pd and Pd-Au/SiO2 catalysts [J].
Applied Catalysis A, General, 2002, 229: 117-125.
[55] Kondratenko E V, Buyevskaya O, Baerns M. Mechanistic insights in the activation of oxygen on oxide catalysts
for the oxidative dehydrogenation of ethane from pulse experiments and contact potential difference measurements [J]. Journal of Molecular Catalysis. A, Chemical, 2000,158:199-208.
[56] 张立德,牟季美. 纳米材料和纳米结构[M]. 北京: 科学出版社, 2001. 413-450.
[57] 董守安. 纳米技术中的金元素. 贵金属, 2003, 24(1): 54-61.
[58] Okumura M,Coronado J M,Soria J, et al. IEPR study of CO and O2 interaction with supported Au catalysts [J].
Journal of Catalysis, 2001, 203: 168-174.
[59] Markus M Schubert Stefan Hackenberg, Andre C van Veen,Martin Muhler, et al. CO oxidation over supported
gold catalysts-"inert" and "active" support materials and their role for the oxygen supply during reaction [J].
Journal of Catalysis, 2001, 197: 113-122.
[60] Hao Zh, Fen L, Lu G Q, et al. In situ electron paramagnetic resonance (EPR) study of surface oxygen species on
Au/ZnO catalyst for low-temperature carbon monoxide oxidation [J]. Applied Catalysis, A, General, 2001, 213: 173-177.
[61] Tripathi A, Kamble V, Mgupen N Microcalorimetry, Adsorption and reaction studies of CO,O2 and CO+O2 over
Au/Fe2O3,Fe2O3 and polycrystlline gold catalysts [J]. Journal of Catalysis, 1999, 187: 332-342.
(上接第27页)
[2] 胡昌义,李靖华,高逸群等. CVD在铱涂层和薄膜制备中的应用[J].贵金属,2002,23(1);53-56.
[3] Dwyer F P, Sargeson A M. The preparation of tris-acetylacetone–rhodium(Ⅲ) and –iridium(Ⅲ) [J]. J. Am. Chem.
Soc., 1953, 75: 984 –985.
[4] Walter Richard, Franz Renate. Preparation of tris(acetylacetonato) Iridium–(Ⅲ) [P]. Eur. Pat.: 1 088 812, 1999.
[5] Gibson D. Carbon-bonded beta-diketone complexes[J]. Coord. Chem. Rev.,1969, 4: 225-240.
[6] 黄德如, 汪仁庆. 无机和配位化合物的红外和拉曼光谱[M]. 北京: 化学工业出版社,1986.256-258.
[7] 周名成, 俞汝勤. 紫外与可见分光光度分析法[M]. 北京: 化学工业出版社,1986.
纳米金催化剂参与的反应
纳米金催化剂参与的反应 2016-05-04 12:46来源:内江洛伯尔材料科技有限公司作者:研发部 纳米金催化剂参与的 反应 纳米金用途广泛,但在当下的生活中,纳米金主要用于催化如下反应: (1) CO 催化氧化 降低燃料电池成本有效方法之一是利用甲醇重整产生的富氢气体。通常该混合物中含 75 %氢气、24 %二氧化碳和 1 %一氧化碳。CO 的存在会导致 Pt 催化剂中毒,因此需要除去 CO,而对 CO 选择性氧化是一种有效方法。同时,CO 低温(常温) 催化氧化过程,涉及空气净化、封闭式 CO2激光器、CO 传感器、防毒面具等多个 方面。目前使用的催化剂的缺点或者是稳定性太差,或者对毒物太敏感,或者反应过程中放出氯化氢造成二次污染。负载型 Au 催化剂,显示出较强的催化氧化 CO 活性和较弱的催化氧化 H2的活性,以及其它催化剂所无法比拟的抗硫中毒能力。(2)水煤气变换反应 鉴于聚合物电解燃料电池在汽车和居民电热传输系统的应用前景,近年来低温水煤气变换反应再度引起国内外学者的兴趣。与己经商业化的 Ni、Cu 基催化剂(其使用温度分别为 900 K或 600 K)相比,负载型金催化剂的使用温度低(473 K)。 (3)选择性加氢反应 Okumura等报道丁二烯在 Au/Al2O3 催化剂上选择性加氢生成丁烯,选择性为 100 %。同时,碳氧化物催化加氢反应生成甲醇是一个重要的化工过程。 (4)选择性氧化有机反应 Onal等报道了在催化氧化 D-葡萄糖成 D-葡萄糖酸反应中,在反应温度为323 K,p H 值为 9.5,Au/活性炭为催化剂时,D-葡萄糖酸的产率(83 %)最大。金粒径对催化活性影响很大,金粒子越小,反应速度越快,产率越高。 (5)乙炔氢氯化反应
金属纳米晶体的表面与其催化效应
金属纳米晶体的表面与其催化效应 沈正阳 (浙大材料系1104 3110103281) 摘要:概括纳米材料的表面与界面特性,从金属纳米晶体表面活性与结构介绍其的催化性能,简要概述金属纳米晶体形状与晶面的关系以及金属纳米晶体的成核与生长。 关键词:纳米金属;表面活性;催化;高指数晶面 1.纳米材料的表面与界面 纳米微粒尺寸小,表面能高,位于表面的原子占相当大的比例。由于表面原子数增多,原子配位不足及高的表面能,使这些表面原子具有高的活性,极不稳定,很容易与其他原子结合。强烈的表面效应,使超微粒子具有高度的活性。如将刚制成的金属超微粒子暴露在大气中,瞬时就会氧化,若在非超高真空环境,则不断吸附气体并发生反应。[1] 纳米晶体是至少有一个维度介于1到100纳米之间的晶体。纳米材料主要由晶粒和晶粒界面2部分组成,二者对纳米材料的性能有重要影响。纳米材料微观结构与传统晶体结构基本一致,但因每个晶粒仅包含着有限个晶胞,晶格点阵必然会发生一定程度的弹性畸变,其内部同样会存在各种缺陷,如点缺陷、位错、孪晶界等。纳米金属粒子的形状、粒径、颗粒间界、晶面间界、杂质原子、结构缺陷等是影响其催化性能的重要因素。纳米材料中,晶界原子质量分数达15%~50%,晶界上的原子排列极为复杂,尤其三相或更多相交叉区,原子几乎是自由的、孤立的,其量子力学状态和原子、电子结构已非传统固体物理、晶体理论所能解释。金属纳米晶体研究中,发现面心立方结构纳米金属如 Al、Ni、Cu 和密排六方结构Co都存在孪晶和层错缺陷,Cu纳米金属中存在晶界滑移。 2.金属纳米晶体的催化性能 近年来,关于纳米微粒催化剂的大量研究表明,纳米粒子作为催化剂,表现出非常高的催化活性和选择性。这是因为纳米微粒尺寸小,位于表面的原子或分子所占的比例非常大,并随纳米粒子尺寸的减小而急剧增大,同时微粒的比表面积及表面结合能迅速增大。纳米颗粒表面原子数的增加、原子配位的不足必然导致了纳米结构表面存在许多缺陷。从化学角度看,表面原子所处的键合状态或键
纳米载体的限域效应对催化性能影响机制的研究进展
纳米载体的限域效应对催化性能影响机制的研 究进展 Company Document number:WUUT-WUUY-WBBGB-BWYTT-1982GT
纳米载体的限域效应对催化性能影响机制的研究进展自上世纪末以来, 纳米科学和技术有了长足的进展,其中纳米材料的一个重要特性是,将体系的尺寸减小到一个特定的范围(如 1~100 nm)时,在不添加任何其他组分的情况下,纳米体系的电子结构会发生变化。量子力学已经证明,大量原子组成的固体材料的价电子为连续的“能带”,当这类体相材料在某一方向上被缩小,特别是缩小到纳米尺度时,电子在该方向的运动就受到空间的束缚和限域,这种限域效应将会改变电子运动特性、导致体系电子结构特别是价电子结构的改变,从而可能会产生量子突变。这种体系尺寸对电子特性的调变为催化剂的催化特性进行调控提供了一种很好的途径[1]。. 近几年,部分研究团队在利用纳米材料的限域效应对催化剂的改性以及催化过程的研究等方面开展了创新性的研究工作,并且大量具有影响力的研究报道和文章被发表出来,其中中国科学院大连化学物理所包信和院士团队在这方面的工作开展的较早也很突出。该团队在铂金属颗粒表面加载了过渡金属氧化物,制备出了具有界面限域效应的TMO/Pt非均相逆催化剂(Oxide-on-Metal Inverse Catalysts),利用界面限域效应对催化体系结构和电子特性的影响作用,改善了在催化过程(特别是在催化氧化反应)中传统非均相催化剂容易出现的催化活性中心的失活以及催化功能的失效等问题[2]。 图1两种金属催化体系的结构示意图 (A)传统的氧化物作为载体的金属催化体系(Oxide supported metal system) 和 (B)过渡金属纳米氧化物倒载型催化体系(oxide-on-metal system)
纳米催化剂
纳米催化剂
纳米催化剂进展 中国地质大学,材化学院,武汉430000 摘要:简要介绍了纳米催化剂的基本性质、其相对于其他催化剂的优势,并较详细地介绍了纳米催化剂类型、部分应用以及相对应类型催化剂例子的介绍,以及常见的制备方法及其表征手段,最后介绍了部分国内和国外纳米催化剂的应用,并对其发展方向进行一定的预测。 关键词:纳米催化剂应用制备催化活性进展 近年来, 纳米科学与技术的发展已广泛地渗透到催化研究领域, 其中最典型的 实例就是纳米催化剂(nanocatalysts—NCs)的出现及与其相关研究的蓬勃发展。NCs具有比表面积大、表面活性高等特点, 显示出许多传统催化剂无法比拟的优异特性;此外, NCs还表现出优良的电催化、磁催化等性能,已被广泛地应用于石油、化工、能源、涂料、生物以及环境保护等许多领域。本文主要就近年来NCs 的研究进展进行了综述。 1.纳米催化剂的性质 1.1表面效应 通常所用的参数是颗粒尺寸、比表面积、孔径尺寸及其分布等,有研究表明,当微粒粒径由10nm减小到1nm时, 表面原子数将从20%增加到90%。这不仅使得表面原子的配位数严重不足、出现不饱和键以及表面缺陷增加, 同时还会引起表面张力增大, 使表面原子稳定性降低, 极易结合其它原子来降低表面张力。此外,Perez等认为NCs的表面效应取决于其特殊的16种表面位置, 这些位置对外来吸附质的作用不同, 从而产生不同的吸附态, 显示出不同的催化活性。 1.2体积效应 体积效应是指当纳米颗粒的尺寸与传导电子的德布罗意波长相当或比其更小时, 晶态材 料周期性的边界条件被破坏, 非晶态纳米颗粒的表面附近原子密度减小, 使得其在光、电、声、力、热、磁、内压、化学活性和催化活性等方面都较普通颗粒相发生很大变化,如纳米级胶态金属的催化速率就比常规金属的催化速率提高了100倍。 1.3量子尺寸效应 当纳米颗粒尺寸下降到一定值时, 费米能级附近的电子能级将由准连续态分裂为分立能级, 此时处于分立能级中的电子的波动性可使纳米颗粒具有较突出的光学非线性、特异催化
负载型镍催化剂的制备
负载型镍催化剂的制备文稿归稿存档编号:[KKUY-KKIO69-OTM243-OLUI129-G00I-FDQS58-
科技论文检索与写作作业 ——负载型镍催化剂的制备 一、制备的目的和意义 1.了解并掌握负载型金属催化剂的原理和制备方法。 2.制备一种以金属镍为主要活性组分的固体催化剂。 意义:催化剂在现代化学工业中占有重要地位。镍基催化剂是一种常用的经典催化剂,具有催化活性高、稳定性好和价格较低等优点,已被广泛应用于加氢、脱氢、氧化脱卤、脱硫等转化过程。 二、制备方法、 1.一种负载型镍催化剂的制备方法,其特征在于,具体包括如下步骤:(1)按钛酸丁酯与无水乙醇体积比为1:1.5~1:3的比例将钛酸丁酯与无水乙醇混合,强力搅拌后得到混合溶液,按无水乙醇与醋酸的体积比为10:1~30:1的比例在混合溶液中加入醋酸形成溶液A;(2)按去离子水与无水乙醇的体积比为1:5~1:10的比例将去离子水与无水乙醇混合得到混合溶液,在混合溶液中加入稀盐酸或稀硝酸调节混合溶液的pH为2~5得到溶液B;(3)按溶液B与溶液A的体积比为1:1~1:4的比例将B溶液加入到A溶液中,然后按钛酸丁酯和十六烷基三甲基溴化铵的摩尔比为1:0.05~1:0.3的比例加入十六烷基三甲基溴化铵形成钛溶胶;(4)按γ?Al2O3和钛酸丁酯的摩尔比为1:0.05~1:0.8的比例在步骤(3)中得到的钛溶胶中加入γ?Al2O3,然后按钛酸丁酯与去离子水的体积比为1:0.5~1:2的比例加入去离子水,静置1~5h后干燥、焙烧得到TiO2?Al2O3复合载体;(5)将 TiO2?Al2O3复合载体于浓度为0.05~1mol/L的硝酸镍水溶液中浸渍4~24h,充分搅拌后干燥、焙烧、通氢还原,得Ni/TiO2?Al2O3负载型镍催化剂。
纳米金催化剂及其应用
纳米金催化剂及其应用 摘要:长期以来,黄金一直被视为具有永久价值的“高贵”金属,在人类社会 象征高贵和权力,决定黄金具有这种地位的科学基础是它的化学非活泼性和优良的可加工性。但1989年 Haruta等发现负载在Fe2O3 和 TiO2 等氧化物上的金纳米粒子具有很高低温 CO 催化氧化活性。金催化剂具有其它贵金属不具有的湿度增强效应,在环境污染、燃料电池、电化学生物传感器等方面都有巨大的应用前景,开辟了金作为催化剂的新领域。本文主要纳米金催化剂制备的研究现状及其部分应用。 关键词:纳米金催化剂选择性氧化加氢环境保护 纳米金催化剂的制备: 一、沉积-沉淀法 沉积-沉淀法是将载体浸渍在 HAuCl4 的碱性(pH值为8~10)溶液中,利用带负电荷的金与载体表面间的静电相互作用实现金的沉积。制备的纳米金粒子较好地分散于载体面,但要求载体具有尽可能大的表面积,对制备低负载量 Au 催化剂非常有效。为了获得最大量金沉积,提高金的负载量,整个制备过程对溶液 pH 值有较大的依赖性,溶液的 pH 值决定了金的前体在水中的水解程度,能够直接影响到金在载体上的吸附,当pH值为8~9时,[AuCl(OH)3]-是 HAuCl4 水解产物中吸附能力最强的形式、,但不同的金属氧化物载体其最佳 pH 值有所不同,目前一般将pH值控制在7~10。在沉积-沉淀法中,尿素对控制均匀沉淀非常有效,还可实现金的最大沉积,金负载量可达到12%,但该法仅适用于等电点较高(IEP>6)的 TiO2、Al2O3、CeO2 等载体纳米金的沉积。后来有科学家研究发现,若用浸渍法对表面浸渍吸附了HAuCl4 的催化剂在高温焙烧前用氨水等碱液多次洗涤,同样也可获得与沉积-沉淀法制备的活性相当的金纳米催化剂,这种方法避免了金的流失,克服了沉积-沉淀法受载体等电点限制的缺点。 二、浸渍法 浸渍法被广泛应用于工业制备贵金属催化剂,研究表明,金和载体表面间亲和力比较弱,在制备和反应过程中容易造成金纳米粒子的聚合,使得催化活性降低,通常认为不适合高度分散纳米金催化剂的制备。后来研究发现金催化剂低温催化 CO 氧化中,沉积-沉淀法比浸渍法获得更高活性是因为该法制备过程中
纳米光催化剂研究现状与展望
年月纳米光催化剂研究现状与展望 马成乡 太原学院山西太原030032 摘要:随着水污染环境问题的日益严重,纳米光催化剂的研究也逐渐的开展起来。本文在分析影响纳米光催化剂性能因素的基础上,探讨了纳米光催化剂的研究现状,并对该材料的发展进行了相关探讨。 关键词:纳米光催化剂;影响因素;研究现状 随着我们国家经济的不断发展,生态环境的污染呈现出不断恶化的趋势,各种环境污染事件开始被社会媒体广泛的暴露出来。在种类比较多的环境污染物中,有机物的比例占到了50%以上。其中天然有机物对环境水体的污染比较小,大多数人工有机物对水体环境的污染程度较大。光催化技术与其他治理环境污染的技术相比,并不需要进行二次净化处理,而且这种纳米光催化剂可以循环使用。 一、影响纳米光催化剂的因素研究 影响纳米光催化剂的性能的因素主要体现在以下几个方面:1.催化剂的晶体结构:通常用作光催化剂的TiO 2具有两种晶体结构,分别为锐钦矿型和金红石型。有的研究结构表明,如果在锐钦矿型的晶体上进行金红石型晶体的生产,能够有效的促进锐钦矿型晶体多污染物的吸收。2.纳米催化剂粒径的影响:催化剂粒径的大小对其催化性能具有着比较重要的影响。很多研究结果表明,随着催化剂粒径的降低,光谱能够响应的范围也就越来越广。尤其当光催化剂离子达到纳米级别时,将会具有更高的氧化还原能力。但是随着纳米粒径的进一步减小,光的载流子在表面符合的概率会进一步增加,也就意味着光催化剂性能的下降。3.比表面积的影响:在反应物质比较充足的情况下,表面积越大,催化剂的活性也就越高;另外催化剂表面的活性中心是并不稳定的。 在反应体系与催化剂的反应条件方面主要影响因素表现在以下几个方面:1.反应的温度:一般来说温度对于光子的表面迁移和吸附以及解吸并不会产生比较明显的影响,所以在某种程度上问对对光催化反应的影响比较小。光催化剂在光的作用下进行各类有机物的催化反应过程时,反应速率与温度比较符合阿伦尼乌斯方程的描述。2.溶液PH 值得影响:溶液的PH 值对半导体的能带分布和表面的性质具有较高的影响。徐成杰等人在研究TiO2在降解有机物的过程中发现,当溶液的PH 值为7时,其降解的效率达到最低。3.光强度的影响:当环境中光的强度较低时,降解速率与光照强度程线性关系;中等光照强度,两者呈现平方根线性关系;当进一步增加光照强度时,催化速率的增加并不明显。 二、纳米光催化的掺杂改性以及复合半导体纳米催化剂的研究 当前纳米的光催化性能研究主要集中在TiO 2的光催化剂掺杂改性研究。在很多学者的研究之中,为了进一步减少自由电子与空穴相互复合的概率,可以在二氧化钛中掺杂少量的稀土离子。非金属离子的掺杂可以使得辐射光谱的范围进一步增强,进而可以提高可见光的利用效率。最近十年以来,双组份甚至是多组分掺杂已经成为纳米光催化剂TiO 2改性研究的热点。美国华盛顿大学的S AKATania 等学者采用溶胶凝胶法制备了La-N-TiO 2光催化剂,ES R 实验研究表明,这种经过掺杂改性的催化剂在500-678nm 光源的照耀下,对于乙醛的降解具有优异的效果。 最近几年以来半导体复合光催化剂的研究引起了学者的广泛注意。从本质上来说,半导体复合就是指一种物质粒子对另外一种物质粒子的修饰。目前的研究结果表明复合半导体比单一半导体具有更好的光催化效果。Tang 等人制备了CaIn 2O 4复合半导体,在亚甲基蓝120min 的脱色实验内,其脱色率可以达到96%。T ony 等人研制除了Fe 2O 3-S nO 2、CuO-SnO 2等类型的复合纳米半导体光催化剂。 三、展望 纳米光催化剂对当前环境问题的解决提供了比较合理的方案,但是目前环境中的光催化剂研究还停留在实验室阶段,并没有得到广泛的应用。目前影响纳米光催化性能的因素主要包括了催化剂的晶体结构、比表面积、反应温度、PH 值等因素;其次对纳米光催化的掺杂改性以及复合半导体纳米催化剂的研究现状进行了一定的分析,指出在以后的污水处理方面,应该设计比较简单的工艺组合反应来处理废水中的污染物,使得纳米光催化剂能够真正的从实验室走向社会。 参考文献: [1]GuoX.,Yang J.,Deng Y.et.al Hydrothermal synthesis and photoluminescence of hierarchic al lead tungstate superstructures re f f ects of reaction temperature and surf actanats[J].European Journalof Inorganic Chemistry,2013,2010(11):1736-1742. [2]SeguraPA,Frane oisM,Ga gnonC,etal.Reviewof theoeeurreneeo f anti-inf eetivesin contaminatedwastew atersandnatUr alanddrinkingw a ters[J].EnvironHealthpersP,2012,117(5):675-684. 管理创新 2014129
纳米金催化剂及其应用
纳米金催化剂及其应用 一.纳米金催化剂的发展 早在1972年,Bond在一篇综述中就指出,第Ⅷ族金属,特别是钯、铂的催化活性都要远高于金的催化活性。金属催化剂主要使用第Ⅷ和ⅠB族的12个金属。用得最多的是3d金属元素Fe、Co、Ni、Cu,4d金属元素R h、Pd、Ag,以及5d金属元素Pt。因此在选用催化剂活性组分的时候,很少在第一时间考虑使用金。1985年Schwank的综述中则这样的评价金的催化剂性:尽管本身不具有反应活性,但金的存在,能够影响第Ⅷ族金属的活性和选择性。而到1999和2000年,Bond和Thompson就金的催化行为相继发表综述性的文章。这足以证明,金已经被作为一种具有优异催化性能的金属元素来使用。特别是在一些多相或者均相反应中,金的催化活性和选择性引起了人们的广泛注意。而这个有无到有、到丰富的过程,仅仅花了15年。在这15年的时间里,大量的研究工作彻底改变了改变了人们对金催化惰性本质的看法。 20世纪80年代中期,关于金催化剂的研究,相继出现了两个突破性进展。1985年发现,英国威尔士大学的Hutching教授,发现纳米金催化剂是催化乙炔氧氯化反应最好的催化剂:1987年,日本学士春田正毅博士发现,负载型纳米催化剂具有低温催化CO的功能。这些研究工作,在当时并没有引起高度重视,但是自从进入20世纪90年代,越来越多的人意识到将纳米金负载在氧化物载体上所产生的新的多相催化行为,对丰富催化剂的制备科学以及催化理论将产生重要影响。 20世纪90年代中期,有关纳米金的研究引起一些国家的注意。在日本美国英国以及意大利等发达国家,集中了相当的人力物力展开此方面的科学研究。有关纳米金方面的研究论文如雨后春笋般见诸各期期刊。关于金催化剂的研究呈现出不断深入逐步扩展的局面。目前,以纳米金作为主题的国际性催化会议,已经举办了三次,也进一步说明,学术界以及产业部门对金的催化作用给予极大的关注,并预示着金催化剂具有不断增长更广泛的应用前景。与此同时,我国在此方面的研究也逐步展开。 二.纳米金催化剂的性质 1.金的物理化学性质 在自然界中,金只以一种稳定的非放射性的同位素形式存在。在任何温度下,空气和氧气对金都不起氧化作用。在所有金属元素中,货币金属属于非稳定的一类,它们的稳定性按电离能力排列为金>铜>银。由于离子半径大,铜银金的金属晶体构型为立方面心晶格,具有熔点沸点高的特点。单组分金属得到的催化剂耐热性差,对使用温度的要求比较苛刻,因此,在工业上为了防止催化剂的失活,要求一定要有适当的助催化剂或载体。 金的熔点汽化热比银要大,较接近铜,这说明金原子之间的键强较强。精确测量表明,金原子金属半径比银稍小。金的电负荷性非常高,只比硫和碘稍稍电正性一点,其亲电子性比氧还强。事实上,金可以一-1价的稳定氧化态存在。另外,进容易于铜铝钛等形成一定组合的合金。 在所有元素中,金的收缩率最大,其半径比没有相对论影响的情况下收缩了15%。金的物理化学性质,可能与其特殊的6s价的电子的半径有关。由于6s价的电子的束缚能被加强,因此导致金很高的电负性和化学惰性。 2.金的催化特性 金的第一电离能力很大,很难失去电子,因此金与表面分子之间的互相作用力通常是很弱的。在低于200℃的温度下,在单晶金的表面,连极具反应活性的分子,如氢氧等,都不易吸附。由于分子在催化剂表面的吸附是催化反应的先决条件,因此可以认为单质金对氢化反应和氧化反应不具有很好的活性。金不具有很好的催化活性,事实上,金催化剂具有催化活性的前提是制备得到高分散的纳米级的金粒子。 3.纳米金粒子的吸附作用 传统方法制备的负载型金催化剂,活性较差,主要是因为它不像其它贵金属催化剂一样高分散。而现在制备得到的粒径在3mm-10mm的纳米催化剂,则显示了特别的优异的催化活性。 纳米粒子是指粒子尺寸为纳米数量级的超细粒子,它的尺寸大于原子簇,小于普通的粒子。纳米粒子是由有限数量的原子或分子组成的,是保持原来物质化学性质并处于亚稳态的原子团或分子团。纳米粒子的表面原子所处的的晶体场环境及结合能与内部原子有所不同,存在许多悬空键,具有不饱和的性质,因而极易与其它原子相结合,所以,具有很高的化学活性,同时也容易吸附其它原子发生化学反应。这种表面原子的活性,不但引起纳米粒子表面构型的变化,同时,任何发生在表面的化学反应,都会因为纳米粒子的存在而表现不同。 随着粒径的减小,金催化剂表面的化学吸附及反应活性相比块体金出现了明显变化:①表面原子的比
纳米催化剂的介绍及其制备
纳米催化剂的介绍及其制备 --工业催化剂小论文 姓名:蒋应战 班级:化工091 学号:0806044111(32号) 指导老师:宫惠峰老师 学校:邢台职业技术学院
目录 1.纳米材料作催化剂的特点 (2) 2.纳米催化剂制备……………………………….. ..2-3 3.微乳液法制备纳米催化剂………………………...4-9 4.纳米粒子催化剂的应用 (10) 5.纳米催化剂的展望................................. . (11) 参考文献................................. . .. (11)
纳米催化剂的介绍及其制备 纳米材料是指颗粒尺寸为纳米量级(1nm~l00nm)的超细粒子材料。纳米技术是当前材料学中研究的前沿和热点,纳米粒子具有比表面积大、表面晶格缺陷多,表面能高的特性,在一些反应中表现出优良的催化性能。纳米催化剂的制备已成为催化剂制备学科中的一个热点。纳米催化剂相对常规尺寸的催化剂具有更高的表面原子比和比表面积,其催化活性和选择性大大高于传统催化剂,可作为新型材料应用于化工中。 1. 纳米材料作催化剂的特点 工业生产中的催化剂应具有表面积大,稳定性好,活性高等优点。而纳米材料恰恰满足这些特点。采用纳米材料制备的催化剂比常规催化剂的催化效率选择性更高。例如,利用纳米材料可用作加氢催化剂,粒经小于0.3nm的镍和铜—锌合金的纳米材料的催化效率比常规镍催化剂高10倍。又如纳米稀土氧化物/氧化锌可作为二氧化碳选择性氧化乙烷制乙烯的催化剂,用这种纳米催化剂,乙烷和二氧化碳反应可高选择性地转化为乙烯,乙烷转化率可达60%,乙烯选择性可达90%。 1.1 纳米催化剂的表面与界面效应 纳米催化剂颗粒尺寸小,位于表面的原子占的体积分数很大,产生了相当大的表面能,随着纳米粒子尺寸的减少,比表面积急剧加大,表面原子数及所占的比例迅速增大。例如,某纳米粒子粒径为5nm时,比表面积为180/g,表面原子所占比例为50%,粒径为2nm时,比表面积为450/g,表面原子所占比例为80%,由于表面原子数增多,比表面积大,原子配位数不足,存在不饱和键,导致纳米颗粒表面存在许多缺陷,使其具有很高的活性,容易吸附其它原子而发生化学反应。这种表面原子的活性不但引起纳米粒子表面输送和构型的变化,同时也引起表面电子自旋、构象、电子能谱的变化。 1.2纳米催化剂的量子尺寸效应 当粒子的尺寸降到(1~10)nm时,电子能级由准连续变为离散能级,半导体纳米粒子存在不连续的最高被占据分子轨道和最低未被占据的分子轨道能级,能隙变宽,此现象即量子尺寸效应,量子尺寸效应会导致能带蓝移,并有十分明显的禁带变宽现象,使得电子/空穴具有更强的氧化电位,从而提高了纳米半导体催化剂的光催化效率。 1..3纳米粒子宏观量子隧道效应 量子隧道效应是从量子力学观点出发,解释粒子能穿越比总能量高的势垒的一种微观现象。近年来发现,微颗粒的磁化强度和量子相干器的磁通量等一些宏观量也具有隧道效应,即宏观量子隧道效应。研究纳米这一特性,对发展微电子学器件将具有重要的理论和实践意义。 2. 纳米催化剂制备 目前制备纳米材料微粒的方法有很多,但无论采用何种方法,制备的纳米粒子必须符合下列要求:a.表面光洁;b.粒子形状、粒径及粒度分布可控;c.粒子不易团聚、易于收集;d.包产出率高。
负载型镍催化剂的制备
科技论文检索与写作作业 ——负载型镍催化剂的制备 一、制备的目的和意义 1. 了解并掌握负载型金属催化剂的原理和制备方法。 2. 制备一种以金属镍为主要活性组分的固体催化剂。 意义:催化剂在现代化学工业中占有重要地位。镍基催化剂是一种常 用的经典催化剂,具有催化活性高、稳定性好和价格较低等优点,已被广泛应用于加氢、脱氢、氧化脱卤、脱硫等转化过程。 二、制备方法、 1.一种负载型镍催化剂的制备方法,其特征在于,具体包括如下步骤:(1)按钛酸丁酯与无水乙醇体积比为1:1.5~1:3的比例将钛酸丁酯与无水乙 醇混合,强力搅拌后得到混合溶液,按无水乙醇与醋酸的体积比为 10:1~30:1的比例在混合溶液中加入醋酸形成溶液A;(2)按去离子水与无 水乙醇的体积比为1:5~1:10的比例将去离子水与无水乙醇混合得到混合溶液,在混合溶液中加入稀盐酸或稀硝酸调节混合溶液的pH为2~5得到溶液B;(3)按溶液B与溶液A的体积比为1:1~1:4的比例将B溶液加入到A溶液中,然 后按钛酸丁酯和十六烷基三甲基溴化铵的摩尔比为1:0.05~1:0.3的比例加入 十六烷基三甲基溴化铵形成钛溶胶;(4)按γAl2O3和钛酸丁酯的摩尔比为1:0.05~1:0.8的比例在步骤(3)中得到的钛溶胶中加入γAl2O3,然后按钛酸丁酯与去离子水的体积比为1:0.5~1:2的比例加入去离子水,静置1~5h后干燥、焙烧得到TiO2Al2O3复合载体;(5)将TiO2Al2O3复合载体于浓度为
0.05~1mol/L的硝酸镍水溶液中浸渍4~24h,充分搅拌后干燥、焙烧、通氢还原,得Ni/TiO2Al2O3负载型镍催化剂。 2.一种用于氨分解制氢的负载型镍催化剂,活性组分为Ni,载体为氧化硅、氧化铝或氧化钛;活性组份的质量百分含量为1-50%。其制备步骤为:将可溶性镍盐、pH值调节剂、沉淀剂、载体以及去离子水配成悬浊液;悬 浊液加热至70-110℃沉积60-300分钟;上述悬浮液降至20-30℃后并过滤,水洗涤、过滤;在80-120℃干燥18-24小时,400-900℃焙烧2-6小时;在氢气气氛,或者氢气和氦气的混合气气氛中,于400-900℃活化3-5小时,还 原制成负载型纳米镍催化剂。本发明催化剂对氨分解反应具有较高的活性,可以应用于氨分解制不含COx氢气的工艺,还可用于各种含氨气体的净化处理过程。 3.一种用于浆态床甲烷化负载型镍基催化剂重量百分比组成为: NiO10-40wt%;载体56-90wt%;助剂为0-4wt%。配制浓度为0.5~1.3g/ml 的硝酸镍与助剂的可溶性盐溶液,依次向其中加入催化剂载体和可溶性有机燃料,搅拌条件下浸渍6-24h,浸渍结束后将溶液于60-90℃水浴条件下加 热浓缩,或直接在300-700℃加热点燃,将燃烧后余下粉末收集,研磨,造粒,在固定床500-700℃用还原气进行还原2-6h,即得到负载型镍基催化剂。本发明具有浆态床甲烷化工艺,且催化性能稳定好,可大规模工业化的优点。 4.一种用于α-蒎烯加氢反应负载型镍催化剂的制备方法和应用,该负 载型镍催化剂的制备工艺步骤包括:在钛酸丁酯中加入无水乙醇后强力搅拌,然后加入醋酸,充分搅拌形成溶液A;将去离子水与无水乙醇混合后调节pH 值得到形成溶液B;把B溶液滴加到A溶液中,加入十六烷基三甲基溴化铵
金催化剂及其在化工中的应用研究进展
金催化剂及其在化工中的应用研究进展在很长时间内,金元素因具有高度稳定性而都被认为是化学惰性的。自从1989年研究人员发现负载在过渡金属氧化物上的金催化剂对CO低温氧化表现出很高的催化活性之后,金催化剂引起了人们的极大兴趣与关注。由于黄金的价格远远低于铂和钯的价格,而且其价格比较稳定,因此开发和研究金催化剂具有明显的经济优势。自1990年以来,有关金催化剂的研究和开发日益活跃。国内研究人员先后论述了2002~2003年金催化剂在有机反应中的研究进展。近几年来,金催化剂在许多新的反应中取得了一定的研究成果,如甲醇部分氧化制氢和苯乙烯环氧化等,预示金催化剂的研究和开发将不断扩大。本文主要介绍了2003年以来金催化剂的制备及其在化工中的应用研究进展,并分析了今后的研究重点。 1 影响金催化剂活性的因素 1.1 制备方法的影响 金的催化活性是通过采用一定的制备方法将金负载在载体上而得到体现的。目前,制备金催化剂的方法很多,常用的方法主要有:浸渍法、共沉淀法和沉积-沉淀法。采用不同方法制备的金催化剂,催化活性往往差异较大。 传统浸渍法是将载体浸渍于氯金酸水溶液中,然后经过干燥、焙烧处理得到金催化剂。使用该方法制备的金催化剂由于富含氯离子而容易形成较大的金晶粒,并且分散性很差,难以得到高活性的负载型金催化剂。 共沉淀法是将HAuCl4水溶液与相应载体的硝酸盐溶液,在一定的碱溶液中进行沉淀,然后经过滤、洗涤、干燥和焙烧得到金催化剂。使用这种方法制备的金催化剂,如Au/α-Fe2O3,在-73℃下对催化氧化CO就具有良好的低温活性。 沉积-沉淀法是将载体悬浮在一定浓度的氯金酸水溶液中,采用碱液调节溶液的pH值,使金物种以氢氧化金的形式沉积在载体表面。如果悬浮液的pH值调节适当,金物种则以非常小的晶粒高度分散在载体表面,得到的金催化剂具有很好的低温活性,从而可以减少氯金酸的用量,提高金的利用率。 由于采用共沉淀法和沉积-沉淀法制备金催化剂的过程中,得到的金催化剂前体往往经过多次过滤和洗涤,可以将吸附在载体表面的氯离子去除,从而明显地减少氯离子对金催化剂的毒害作用,提高金催化剂的活性,因此用共沉淀法和沉积-沉淀法比浸渍法更加可取。共沉淀法与沉积-沉淀法相比,有研究认为后者更优于前者,因为采用
大肠杆菌合成金纳米粒子复合催化剂性能研究
大肠杆菌合成金纳米粒子复合催化剂性能研究 2016-08-01 13:16来源:内江洛伯尔材料科技有限公司作者:研发部 Au@TiO2催化剂的TEM照片自上世纪八十年代Hutchings和Haruta等发现金催化剂具有高催化活性以来, 金催化剂的研究受到密切关注, 目前已取得很大进展. 但金催化剂很少用于工业应用. 原因之一是由于金粒子的聚集长大及表面碳酸盐物种的积累而导致金催化剂易于失活. 如何有效阻止金粒子的聚集, 提高金催化剂的稳定性已成为目前亟待解决的问题. 近年来, 金属纳米粒子与DNA、蛋白质、壳聚糖等生物大分子的相互作用及其自组装研究引起人们的密切关注. Baron等评述了以DNA、蛋白质等生物分子为模板合成Au、Ag纳米粒子和纳米线的研究进展. 这种材料既可以通过生物分子的识别和催化功能来改善金属纳米粒子的电学、光学和催化性能, 也可以通过改性金属纳米粒子来改善生物分子的某些性能. Horovitz等发现柠檬酸钠还原的金纳米粒子与大麦糊粉层细胞提取的蛋白质之间存在静电作用. 杨芳等研究了藻蓝蛋白对Au3+离子的原位还原和纳米Au0形成的动态过程, 发现藻蓝蛋白的紫
外特征吸收峰强度随Au3+离子浓度的增加和放置时间的延长而降低, 其荧光发射峰和荧光激发峰也呈现衰减趋势, 提出藻蓝蛋白中的半胱氨酸、胱氨酸和色氨酸可将Au3+还原为Au0. 金明善等研究了金纳米粒子和R-藻红蛋白的相互作用, 发现R-藻红蛋白对金纳米粒子有良好的稳定作用. Huang等发现壳聚糖能保护金纳米粒子. 刘克增等制备了金@壳聚糖复合材料, 发现该材料对葡萄糖空气氧化制葡萄糖酸具有良好的催化性能.另一方面, 微生物与金属纳米粒子的研究也日益增多. Gericke等详细评述了各种微生物在制备金纳米粒子方面的研究进展, 认为可以通过调变微生物的生长参数(如培养时间、pH 值、温度等)达到对金纳米粒子形貌和尺寸的控制. 某些菌体如枯草芽孢杆菌、酵母菌、真菌等能够聚集并还原金离子, 已用于金纳米粒子和纳米线的合成. 研究表明, 细胞中的羟基和氨基可作为Au3+的结合位, 而醛基可作为电子供体将Au3+还原成Au0. Kuo等利用大肠杆菌对金离子的还原作用制备了金@大肠杆菌复合材料, 发现这种材料具有很强的生物相容性,可望应用于光热治疗癌细胞方面. 傅锦坤等用细菌将Au/α-Fe2O3上的Au3+还原成Au0, 焙烧后获得的催化剂与浸渍法制备的催化剂相比有较高的CO氧化反应活性.可以看出,目前的研究主要集中于微生物对金属离子的吸附与还原作用以及金属纳米粒子的制备, 而将其用于催化领域的报道较少. 鞭毛是细菌表面的运动器官, 由单一的鞭毛蛋白组装形成螺线管状结构, 鞭毛的长短和数量可以通过改变细菌的培养条件来调控. 最近, Kumara等首次实现了Au、Ag、Cu 等金属纳米颗粒在细菌鞭毛表面的组装. 利用细菌鞭毛为模板制备二氧化钛等无机氧化物纳米管也已获成功. 但尚未见利用此法制备金催化剂的研究. 大肠杆菌为革兰氏阴性短杆菌, 为杆状结构, 具有抵抗力强、易培养等优点. Nomura等以大肠杆菌为生物模板合成了氧化硅的空心纳米管. 烟台大学化学生物理工学院索掌怀等人利用大肠杆菌(DH5α)对金属离子较强的吸附与还原能力制备了Au@DH5α, 再利用大肠杆菌的水分来水解钛酸四丁酯, 得到Au@DH5α -Ti(OH)4样品, 焙烧去除大肠杆菌后得到氧化钛包裹的纳米金粒子催化剂Au@TiO2. 以N2吸附,
纳米金
摘要:纳米金催化剂具有高催化活性和选择性,作为新型催化材料引起关注。尝试用胶体浸渍法 将金催化剂负载于基体材料上,以解决纳米金颗粒难于均匀负载于基体材料表面等问题,并重点对纳米金催化剂的应用进行了评述。 金历来被认为呈催化惰性,但20世纪80年代HarutaM等开创性地发现,负载于氧化物上的纳米金催化剂在CO室温氧化中表现出非常高的反应活性,纳米金作为新型催化材料引起关注,其应用涉及污染治理、化工过程和H的开发与利用(如燃料电池、选择性氧化CO、水蒸汽变换反应)等方面。纳米金催化剂显著特征是低温活性,许多催化反应都可在室温下实现高活性催化,有些反应甚至可以在0℃实现完全转化,可见金催化剂具有非常低的表观活化能;金催化剂具有好的选择性,Au/Al2O3催化剂催化丁二烯加氢反应可100%生成丁烯;同时,金催化剂比铂族催化剂廉价。本文介绍纳米催化剂的制备方法,并重点对纳米金催化剂的应用进行评述,旨在为纳米金催化剂的应用开发提供参考。1纳米金催化剂的制备 金催化剂制备方法主要是浸渍法、沉积沉淀法和共沉淀法。浸渍法虽然被广泛用于工业制备贵金属催化剂,但许多研究表明,该法不适合于金催化剂的制备,主要是因为制备的金催化剂分散性不好,金颗粒大。共沉淀法和沉积沉淀法是金催化剂制备的常用方法,但共沉淀法的最大缺陷是所需负载量大(一般认为纳米金颗粒被载体包裹,有效活性部位减少)。而沉积沉淀法解决了这个问题,制备的纳米金粒子较好地分散于载体表面,但要求载体具有尽可能大的表面积,整个制备过程对溶液pH有较大的依耐性,当pH为8~9时,[AuCl(OH)3]-是HAuCl4水解产物中吸附能力最强的形式,因此,为获得最大量金沉积,应将pH控制在8~9,沉淀剂的选择直接影响催化剂制备过程中pH的变合处理低浓度的CO。相比这些催化剂,金催化剂显化,使用的沉淀剂是NaOH和Na2CO3,采用Na2CO3具有良好的低温催化氧化CO活性,抗水性能好,比铂和钯催化剂廉价。可避免引入杂质Na+;用NaOH作沉淀剂时,溶液pH不稳定,而且金的沉积量也有限。文献报道,在沉积沉淀法中,尿素控制均匀沉淀过程非常有效,可以实现金的最大沉积。 年来,由于不同的实验需求,许多研究者开发出一些新的制备方法。IvanovaS等开发出阴离子交换法(DirectAnionicExchange,DAE),其原理是利用金络合物的OH基团与载体表面的OH基团发生置换反应,将金以氢氧化金的形式负载于载体表面。以Al2O3作载体,用DAE法制备Au/Al2O3催化剂,实验发现,不同HAuCl浓度对制备的催化剂活性有较大影响,浓度越低,负载效果越好,制备的催化剂活性越高。不同的洗涤方法对催化剂活性也有较大影响,氨水洗涤比水洗的催化活性好,因为氨水水解产生的OH与Cl发生交换,减少了催化剂表面Cl的残存。DomínguezMI等用混合氧化物制备的泡沫作基体材料,负载一层CeO后,用DAE法负载金,用于CO氧化。虽然这种制备方法操作简易,但负载的金沉积量有限,很难应用于一些需较高金沉积量的催化反应。MallickK等开发了一种新型简易金催化剂制备方法———硼氢化钠还原法。将载体氧化物悬浮于蒸馏水中,剧烈搅拌,滴入HAuCl溶液,静置,剧烈搅拌下,加入NaBH4燥,得催化剂。实验发现,该法制备的Au/TiO2催化溶液,老化,过滤,洗涤,干燥,得催化剂。实验发现,该法制备的Au/TiO催化 剂比其他方法制备的Au/TiO2催化剂具有更高的CO氧化活性。除以上几种制备方法外,用于金催化剂制备的法还有化学气相沉积法、有机金配合物固载2.2丙烯环氧化法、光化学沉积法和直流磁电管溅射法等,相比前面介绍的几种制备方法,这些制备方法对仪器设备或对金前身化合物物性要求较高。
金纳米催化剂(八):其他类型
金纳米催化剂(八):其他类型 2016-08-22 13:23来源:内江洛伯尔材料科技有限公司作者:研发部 金纳米催化剂 除了聚合物基弹性网络、中空球型、核壳型和蛋壳型纳米Au催化材料,目前还有片状、柱状等各种类型纳米Au催化材料合成的报道。 基于独特的结构性能和电子迁移性质,石墨烯一经出现即在科学界掀起巨大波澜。目前,石墨烯在纳米Au催化材料定向合成中同样受到广泛关注,并取得了丰硕研究结果。例如,Lu 等利用TWEEN 20 同时作为氧化石墨烯(GO)的稳定剂和AuNPs的还原剂,通过原位还原HAuCl4获得AuNPs/TWEEN/GO片状复合材料。催化NaBH4还原4-NP的反应发现,由于GO的协同效应,复合材料呈现良好催化活性:催化同一反应,AuNPs/TWEEN/GO可在14 min 完成反应,拟一级速率常数为25.37 ×10-2s-1,而AuNPs/ TWEEN用时32 min,拟一级速率常数为9.05×10-2 s-1。. Chen等以NaBH4为还原剂,制备了具有热响应催化行为的片状石墨烯(GO)负载的AuNPs(GO-PNIPAAm聚异丙基丙烯酰胺)-Au)。复合材料在催化NaBH4还原4-NP 的反应中表现出温度调控的催化活性:当温度高于PNIPAAm的临界溶解温度时,PNIPAAm链由亲水相转变为疏水相,造成末端AuNPs的团聚,进而导致催化活性降低。较之普通石墨烯基纳米催化剂,其具有可调控的催化行为。
Li等采用三乙烯四胺为还原剂,通过水热体系中AuNPs与石墨烯的自组装过程成功合成了高1.28cm和直径1.08 cm的圆柱形Au/石墨烯复合材料。基于石墨烯的结构性能,其在室温催化NaBH4还原4-NP的反应中呈现优异催化活性,12 min反应基本完成,拟一级速率常数为3.17 ×10-3 s-1。分别高出先前文献报道的海绵状和高聚物负载纳米Au复合材料的催化活性90倍和14倍。 此外,Zhang等采用单毛细管电纺技术与原位还原技术相结合的方法,将高度分散、尺寸较小的AuNPs成功组装到SiO2纳米管的内、外表面(AuNPs/SNTs)。该管状纳米复合物对NaBH4还原4-NP的反应表现出高催化活性:反应不到5 min即可完成,拟一级速率常数为1.06 ×10-2 s-1。 Jin等采用两步法,将分散的、尺寸可控的AuNPs固载于分层结构的层状硅酸镍(NiSiO)上,获得像花一样的纳米复合物(AuNPs/NiSiO)。该复合物对NaBH4还原4-NP的反应表现出高催化活性:2.5min反应即可完成,拟一级速率常数为1.63×10-2 s-1。且循环使用5次后,活性没有下降,表明该复合物具有良好的稳定性。
金纳米催化剂(四):二氧化硅磁性核壳结构
金纳米催化剂(四):二氧化硅磁性核壳结构 2016-08-22 13:15来源:内江洛伯尔材料科技有限公司作者:研发部 多功能Fe3O4@SiO2-金纳米粒子合成路线2011年,Zhu等以NaBH4为还原剂,制备了多功能Fe3O4@SiO2?AuNPs复合微球。微球饱和磁化强度为23.9 emu/g,尺寸均一,AuNPs稳定存在,且尺寸较小(4nm,TEM)。在NaBH4室温还原4?NP的反应中表现出高催化活性,12min 4?NP的转化率达到95%;催化剂便于磁性回收,具有长效催化寿命和良好的重复使用性,适于规模化制备和实际应用过程。 2013年,Zheng等借助Sn2+修饰Fe3O4@SiO2核壳结构表面并以其作为还原剂制得Fe3O4@SiO2?AuNPs核壳结构催化材料。合成过程简单,不仅减少了合成步骤而且显著降低了合成成本。 磁性复合微球具有高磁性(饱和磁化强度为35.3emu/g)、良好的渗透性和稳定且裸露的AuNPs,AuNPs的分散性较好,平均粒径约5 nm(TEM)。298K时,催化NaBH4还原4?NP的研究结果表明:向反应体系中加入催化剂后,反应快速进行,没有诱导期,4 min反应完成,这在实际应用中是一重要优势;还原产物仅为4?AP,无副产物生成,拟一级速度常数为1.42 ×10-2S-1,比产生相同转化率的Au或Ag基纳米催化剂都要高;反应完成后,催化剂通过外加磁场回收,循环使用前7次转化率稳定在91%,此后转化率稍有下降。该合成路线为制
备其他多功能金属纳米复合物提供了借鉴。同年,Liu等采用表面诱导原子转移自由基聚合(SIATRP)的方法,将聚甲基丙烯酸二甲氨基乙酯(PDMAEMA)接枝到Fe3O4@SiO2@亚甲基双丙烯胺和甲基丙烯酸羟乙酯共聚物(PHEMA)多层微球的表面,用NaBH4原位还原Au3+后制得多壳层结构的Fe3O4@SiO2@PHEMA?g?PDMAEMA?AuNPs核壳型催化材料,AuNPs的尺寸约3.7 nm(DLS)。研究结果表明:合成材料对NaBH4还原4?NP具有良好的催化活性,15 min反应完成,拟一级速度常数为4.5 ×10-3S-1,较Au@meso?SiO2更高。经分离回收、循环使用6次,转化率仅由99.0%下降到96.7%,证实其具有良好稳定性和催化活性。这种将无机过程(溶剂热法、溶胶凝胶法)和有机过程(蒸馏沉淀法、SIATRP)相结合的方法,可方便合成具有特殊结构和高性能的功能材料;然而,由于多壳层的屏蔽作用,无法有效进行磁控分离。 Rahman等制备了具有介孔SiO2(mSiO2)外壳的Fe3O4@SiO2@Au?Shell@mSiO2。制备过程如下:(1)通过溶胶?凝胶法制得Fe3O4@SiO2纳米颗粒;(2)对其表面氨基化并将柠檬酸稳定的AuNPs通过静电作用吸附在氨基化的颗粒表面,以此为种子进一步形成具有Au 壳的复合物Fe3O4@SiO2@Au?Shell;(3)以CTAB为模板剂和TEOS为硅源组装Fe3O4@SiO2@Au?Shell@SiO2核壳型复合物,除去CTAB后获得Fe3O4@SiO2@Au?Shell@mSiO2纳米复合物。复合材料具有高饱和磁化强度(21.4 emu/g),贯穿性良好的介孔SiO2外壳(介孔2.1 nm)及接近性好的Au壳(壳厚25 nm)。室温催化NaBH4还原4?NP的反应,12 min 反应基本完成,拟一级速率常数为2.33 ×10-3 S-1,且速率常数随催化剂加入量增加而增大;反应后经磁控回收、循环使用8次,转化率仍高达90%,表明合成材料具有良好的重复使用性。担载Au的磁性颗粒和介孔壳层所形成的纳米复合材料也可用于定向药物缓释并能强化作用效果。