甾体5α-还原酶抑制剂讲义

Steroidal 5α-Reductase Inhibitors: A Comparative 3D-QSAR Study Review
甾体5α-还原酶抑制剂:一项类比性的三维定量构效关系(3D-QSAR)研究报告Suresh Thareja* (作者姓名)
School of Pharmaceutical Sciences, Guru Ghasidas Central University, Bilaspur, Chhattisgarh 495 009, India
医药科学院,古鲁加思达斯中央大学,比拉斯普尔市,恰蒂斯加尔邦495009,印度
CONTENTS 目录
1. Introduction简介2883
2. Historical Aspect of Published 2D-QSAR Studies 已发布的二维定量构效关系(2D-QSAR)的历史概况2884
3. Current Aspect of 3D-QSAR Studies 三维定量构效关系研究的目前情况2885
4. 3D-QSAR Methodology Employed for Steroidal 5α-Reductase Inhibitors 甾体5α-还原酶抑制剂的三位定量构效关系的研究方法论2885 4.1. Selection of Data Sets and Inhibitory Activities 数据集和抑制活性数据精选2885 4.2. Selection of Training and Test Sets 实验和练习册精选2885 4.3. Molecular Modeling and Alignment 分子建模和校正2886 4.4. 3D-QSAR Models 三维定量构效关系模型2886
4.5. Statistical Analyses 统计学分析2886
5. Results and Discussion of 3D-QSAR Studies on Steroidal 5α-Reductase Inhibitors 甾体5α-还原酶抑制剂的三维定量构效关系研究的结论和研讨2886 5.1. Finasteride Analogues (4-Azasteroids) As Inhibitors of Steroidal 5α-Reductase-II 非那雄胺类似物(4-氮杂类甾醇)作为甾体5α-还原酶-II的抑制剂2886 5.2. Epristeride Analogues (3-Carboxysteroids) As Inhibitors of Steroidal 5α-Reductase-II依立雄胺类似物(3-羧基甾体化合物)作为甾体
5α-还原酶-II的抑制剂2888
5.3. Pregnane Derivatives As Inhibitors of Human Steroidal 5α-Reductase-II孕烷衍生物作为人类的甾体5α-还原酶-II的抑制剂2888
5.4. 6-Azasteroids As Dual Inhibitors of Both Isoforms of Steroidal 5α-Reductase 6-氮杂类甾醇作为甾体5α-还原酶-II的两个亚型的双重抑制剂2888
6. Conclusions结论2890 Author Information 作者信息2892 Corresponding Author 联系人2892 Notes 注解2892 Biography 档案2892 Acknowledgments 特别感谢2892 References 参考文献2892
1. INTRODUCTION 简介
良性前列腺增生(BPH)是由于其机制增生和前列腺的腺体元素造成的非癌前列腺增长。其结果是近端尿道梗阻,从而导致了尿流紊乱①。这是由雄激素双氢睾酮(DHT)的增强水平导致的,这种激素在前列腺生长中起到了重要作用。
甾体5α-还原酶(EC 1.3.99.5)是一种细胞核核膜结合酶,它能在NADPH的辅助下将内源性睾酮(T)转化为双氢睾酮。②DHT在这些组织中刺激细胞增长,因此造成了中老年人体中的前列腺迅速肥大。我们针对使用5α-还原酶抑制从T到DHT的转换过程所提出的化学机理,涉及到了酶和NADPH之间的二元复合物的形成。紧接着的是以T为基层的三元复合物的形成。③甾体5α-还原酶在提升DHT含量的许多情境和恶疾中都扮演着重要角色,包括前列腺增生症,前列腺癌,多毛症,痤疮和男性秃顶。④
甾体5α-还原酶有两种亚型,也就是I型和II型,存在于人类和老鼠的互补DNA(c-DNA)文库中。⑤⑥I型普遍存在于毛囊以及皮肤的皮下腺体中,而II型普遍存在于前列腺、生殖器皮肤、精囊、附睾中。⑦⑧最近,在全基因组基因表达图谱分析的进展下,第三种5α-还原酶(III型)在激素非依赖型前列腺癌细胞中被发现了。⑨⑩
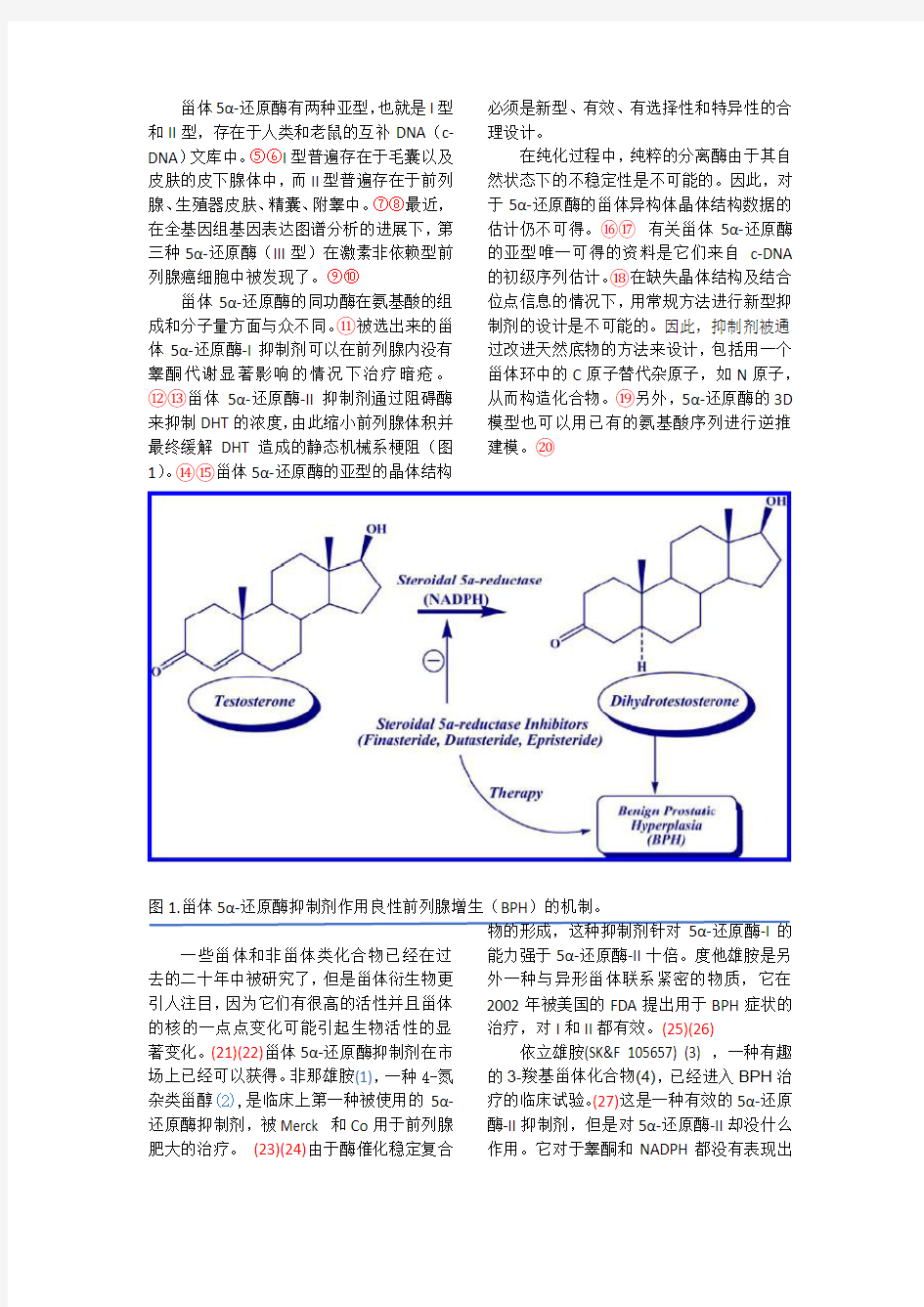
甾体5α-还原酶的同功酶在氨基酸的组成和分子量方面与众不同。?被选出来的甾体5α-还原酶-I抑制剂可以在前列腺内没有睾酮代谢显著影响的情况下治疗暗疮。??甾体5α-还原酶-II抑制剂通过阻碍酶来抑制DHT的浓度,由此缩小前列腺体积并最终缓解DHT造成的静态机械系梗阻(图1)。??甾体5α-还原酶的亚型的晶体结构必须是新型、有效、有选择性和特异性的合理设计。
在纯化过程中,纯粹的分离酶由于其自然状态下的不稳定性是不可能的。因此,对于5α-还原酶的甾体异构体晶体结构数据的估计仍不可得。?? 有关甾体5α-还原酶的亚型唯一可得的资料是它们来自c-DNA 的初级序列估计。?在缺失晶体结构及结合位点信息的情况下,用常规方法进行新型抑制剂的设计是不可能的。因此,抑制剂被通过改进天然底物的方法来设计,包括用一个甾体环中的C原子替代杂原子,如N原子,从而构造化合物。?另外,5α-还原酶的3D 模型也可以用已有的氨基酸序列进行逆推建模。?
图1.甾体5α-还原酶抑制剂作用良性前列腺增生(BPH)的机制。
一些甾体和非甾体类化合物已经在过去的二十年中被研究了,但是甾体衍生物更引人注目,因为它们有很高的活性并且甾体的核的一点点变化可能引起生物活性的显著变化。(21)(22)甾体5α-还原酶抑制剂在市场上已经可以获得。非那雄胺(1),一种4-氮杂类甾醇(2),是临床上第一种被使用的5α-还原酶抑制剂,被Merck 和Co用于前列腺肥大的治疗。(23)(24)由于酶催化稳定复合物的形成,这种抑制剂针对5α-还原酶-I的能力强于5α-还原酶-II十倍。度他雄胺是另外一种与异形甾体联系紧密的物质,它在2002年被美国的FDA提出用于BPH症状的治疗,对I和II都有效。(25)(26)
依立雄胺(SK&F 105657) (3),一种有趣的3-羧基甾体化合物(4),已经进入BPH治疗的临床试验。(27)这是一种有效的5α-还原酶-II抑制剂,但是对5α-还原酶-II却没什么作用。它对于睾酮和NADPH都没有表现出
抑制能力。(28)
因为X射线探测人体甾体5α-还原酶亚型不可行,人们对于在的基础上根据现有抑制剂的结合特性基于配体的药物设计方法设计新型抑制剂有着极大的兴趣。(29)(30)基于配体的药物设计方法利用活性配体相似的事实它更倾向于制造比传统配体更加具有活性的配体。(31)基于配体的方法都考虑了二维和三维化学,空间位阻,静电,和相互作用点(例:药效团点)来进行相似评估。
(32)配体相似性法(2D或3D)只需要一个有活性的的分子,它可能来自于文学,专利,或内部实验数据。这样的话,活动可能只需要一个不是很精确的高通量分析就可以决定。(33) 定量构效关系分析(QSAR)是基于配体用计算机进行研究而进行一系列分子的生物活性和理化性质的方法之一。(34)QSAR 的只要目标是建立最好的药物模型以克服试验误差带来的困难。QSAR也用于降低用于推理药品构造所带来的时间的消耗,因此能提高药品分子的生物活性。QSAR为制造化合物的结构变化提供了指引,因此可以在减少副作用的情况下得到效用更好的药物。
(35)
我们研究小组已经用自建分子场以基于3D-QSAR研究药品的结构框架,从而设计出新的、可选的甾体5α-还原酶抑制剂以用于BPH的治疗和其他相关疾病。(20),(36)-(41)考虑到从初步设计研究中得到的结果,我们在实验室中建立并整理了一个新型化合物库。设计的化合物的抑制活性与我们建立的3D-QSAR所预测的一致。(42)
在这些结果的基础上,一个还可以的3D-QSAR关于不同种类的5α-还原酶抑制剂的研究获得了良好的成效。另外,我们研究的结果能被新型设计和化合物库作为甾体5α-还原酶所接受。
2. HISTORICAL ASPECT OF PUBLISHED 2D-QSAR STUDIES
已发布的二维定量构效关系(2D-QSAR)的历史概况
2D-QSAR技术是非常有趣的,因为他能减少对3D构造的需求,假设结合物的构造,和排列分子。在已有的文献库中,收录了非常多有关甾体5α还原酶抑制剂的2D-QSAR 研究报告。他们的研究结果都描述的非常详尽,而且他们在QSAR描述的基础上提出了有力先进甾体5α还原酶抑制剂的结构构架。
(43)
并且,Hutter 和Hartmann宣布他们有关人体甾体5α还原酶抑制剂的QSAR研究还包括了甾体和非甾体化合物。(44)
有关甾体5α还原酶抑制剂的QSAR研究结论揭示了不同的理化参数造成的不同影响,主要是对摩尔折射度、总偶极矩、氢键供体做出了积极的贡献。同时对巴拉班指数,对数,和最低未占据分子轨道(LUMO)起到了消极作用。(40)于是3D-QSAR模型的建立就成为了必要的,最重要的部分就是分子识别和目标结合。(45)
3. CURRENT ASPECT OF 3D-QSAR STUDIES三维定量构效关系研究的目前情况
三维定量构效关系(3D-QSAR)技术是最合理的基于合理配体的药物设计计算方式。对药物化学家来说,针对于根据分子描述符分析分子分析分子有多重可行的3D-QSAR算法。(46)-(50)一个经验证的3D-QSAR 模型不仅能在更好的理解任何一类分子的构效关系方面起到作用,还能给研究者提供更进一步的分子水平的清晰的洞察力。(51)-(53)
比较分子力场分析法(CoMFA)已经是一种新的基于配体设计药物的立体信息化学规范的工具。(48)自组织分子场分析(SOMFA)也是一种新型的基于场的合理药物设计工具,由Robinson 和他的同事所开发。(54) SOMFA 和CoMFA的相似性让正在发展的3D-QSAR模型成为一种更有吸引力的技术。
(55)-(57)
SOMFA是围绕一套配为体的分子的内部属性(静电和空间位势)取例,并通过将场与相应的有共同靶受体的配位体相关联的实验活动。(58)-(59)这种技术为提供了配体
和受体相互作用的精确的信息。通过这种方法,用3D图谱预知分子的生物学活性和推测分子性质与生物活性之间的关系成为了可能。(60)-(63)
在我们实验室中,SOMFA已经成功地被运用于甾体5α-还原酶,蛋白酪氨酸磷酸酶1B(PTP-1B),二肽基肽酶IV(DPP)、醛糖还原酶(AR)和环氧合酶II(COX-II)抑制剂的设计。
(64)-(67)关于分子结构,不同药效的模型的3D-QSAR研究在数据上有很大的差别,包括4-氮砸类固醇(非那雄胺类似物),3-羧基类固醇(依立雄胺类似物)、孕烷衍生物,和6-氮砸类固醇。(20),(36)-(40)
通过有药效的分子的叠加,,识别其结构特点有助于减弱受体的结合率。(62)而且,该研究产生的空间位阻和静电图形将用于区分两种酶的异构体亚型和键结构的特征。
(68)
总之,准确预测基于结合自由能的未经测试计算的化合物的活性是是复杂而漫长的。批判性的解读SOMFA地图可以导致关键结构特征的识别,可以被利用来提高最有效的参考化合物的效用。(69)
4. 3D-QSAR METHODOLOGY EMPLOYED FOR STEROIDAL 5α-REDUCTASE IN HIBITORS 甾体5α-还原酶抑制剂的三位定量构效关系的研究方法论
4.1. Selection of Data Sets and Inhibitory Activities 数据集和抑制活性数据精选
数据集的广泛的差异构成了4-氮砸类固醇(非那雄胺类似物)(70)(71),3-羧基类固醇(依立雄胺类似物)(72)、孕烷衍生物,(73)-(79)和6-氮砸类固醇(80)-(82)作为甾体5α还原酶抑制剂,与3D-QSAR研究的现状进行对比。这些化合物的抑制能力的数据集已经被来自同一个实验室在每个研究中被选出的单一个体所测量。
为了以线性上升的规律排列数据,我们采取了抑制活性的负对数来研究5α还原酶的亚型。(83)(84)
4.2. Selection of Training and Test Sets实验和练习册精选
为产生一个包括尽可能多的信息的实验册,同时仍旧允许排除化合物,不同子集的化合物被分配到了实验册中。
训练集和测试集的化合物的比例为3:1。数据集被假定分为测试集分子和训练集分子,在测试集的分子结构与类似化合物的训练集类似,根据训练集化合物的测试集的研究,其结论的研究具有较高的代表性和预测能力。(85)
图2. 临床用的非那雄胺(1) 支撑4 -氮杂甾族化合物(2)构成3D-QSAR研究的支架。
4.3. Molecular Modeling and Alignment分子
建模和校正
分子建模是建立一个可靠的3D-QSAR 模型的第一步。所有分子数据集的三维架构都是用Chemdraw Ultra 8.0构造建模的。(86)-(89)在研究中,因为架构有全球最小值,能量最小化被分子力学(MM2)和半经验方法(AM1)所共同结合。汉密尔顿近似值奥斯汀模型1(AM1)被用于缩小架构,以大袋数据集所要求的最小能量值。(90)-(91)
在各种数据集中所有分子的对齐是另外一个最终3D-QSAR模型建设的关键步骤。在3D-QSAR研究中,有关最有效化合物的使用有一个公共的准则被用于目的的一致化。(92)在一些研究中,临床用非那雄胺也使用这种数据集对齐。(36)-(40)
所有的排列结构转换都使用Vega ZZ软件的其他文件格式(93)
4.4. 3D-QSAR Models 三维定量构效关系模型
最初,三维图的40 x40 x40尺寸起源于(-20,-20,-20),被所有训练集分子逐渐包围。
(94)这些图的空间位阻和静电性能都在被使用。
所有的数据集分子,包括测试集的空间位阻和静电势的产生,都使用这些图。(95)红点表示的立体体积大的取代基的存在,而蓝点表示不大的取代基,使复合物与目标更好的互相影响。(64)(66)4.5. Statistical Analyses统计学分析
一个偏最小二乘(PLS)法,多元回归分析的扩展,用于三维定量构效关系的描述,其中SOMFA描述符作为独立变量和抑制活性值因变量。用留一交叉验证(LOO)选项进行最优数量的组件,以用于最终的分析。
在对构建的QSAR模型进行性能评价时,在定量构效关系研究领域有一个常用的方法:Tropsha如下。建议应有预测的QSAR模型的交叉验证相关系数Rcv2(Q2)1范围内的值,表明一个完美的模型误差的预测小于0,而且分配的模型中的每个化合物的活度大于误差。(96)F值越大,表明QSAR模型的统计学概率越大。(97)-(100)
5. RESULTS AND DISCUSSION OF 3D-QSAR STUDIES ON STEROIDAL 5α-REDUCTASE INHIBITORS 甾体5α-还原酶抑制剂的三维定量构效关系研究的结论和研讨
5.1. Finasteride Analogues (4-Azasteroids) As Inhibitors of Steroidal 5α-Reductase-II
非那雄胺类似物(4-氮杂类甾醇)作为甾体5α-还原酶-II的抑制剂
包含非那雄胺类似物(4-氮杂类甾醇)作为甾体5α-还原酶-II抑制剂的数据集的
3D-QSAR研究参考了Rasmusson et al.(70)和Liang et al.(71)被我们小组发表的文章(2010)。
(36)(37)
图3. 为构建的甾体5 -还原酶-II抑制剂而对4-氮杂甾体衍生物中对静电和空间位阻有要求。
图4.临床用的依立雄胺(1)支撑3-羧基类固醇(2)构成3D-QSAR 研究的支架
选择出来的4-氮杂甾体与临床用非那雄胺具有结构相似性(图2)。
最好的模型的统计分析采用PLS 分析,显示出了良好的交叉验证相关系数Q2(0.783),非验证相关系数R2值(0.806),和高F-test 值(87.282),具有良好的相关性
和预测能力。R2周围高密度的蓝点显示负电
基团与甾体5α-还原酶-II 相互作用的需求。
我们的研究显示,复合物对甾体5α-还原酶-II 具有强有力的抑制活动时,一个不大的正电性取代基与R1相关性良好,同时一个大的负电型取代基与R2关系良好。
图5.为构建有力的甾体5 -还原酶-II 抑制剂而对3-羧基类固醇的静电和空间位阻有要求。 5.2. Epristeride Analogues (3-Carboxysteroids) As Inhibitors of Steroidal 5α-Reductase-II 依立雄胺类似物(3-羧基甾体化合物)作为甾体5α-还原酶-II 的抑制剂 Holt et al.(72)声称一系列不饱和的3-羧基类固醇有良好的羧酸盐和带正电荷的氧化辅因子之间的静电相互作用,产生了竞争性的甾体5α-还原酶-II 抑制剂。不饱和3-羧基类固醇与依立雄胺,一种临床用药,有物理结构相似性(图4)。
我们小组研究了一个
3D-QSAR 不饱和
的3-羧基类固醇数据集(2009)。(38)最好的模型统计分析是采用PLS分析,显示了非常好的交叉验证相关系数Q2 (0.693),非验证相关系数R2数值为(0.732),以及高F-test数值(43.816),伴随着良好的相关性和预测能力。
3D-QSAR的立体图显示了高密度的红点在取代R,表示有利空间的相互作用是由于其侧链分支造成的。高密度的蓝点周围的R表示电负性取代基的最佳抑制活性的需求。研究表明一个不大的电负性取代基与C-3和较大的电负性取代基以及Δ(3-4,5-6,11-12)有着和甾体5α-还原酶-II抑制剂活性复合物之间的良好关系。
5.3. Pregnane Derivatives As Inhibitors of Human Steroidal 5α-Reductase-II
孕烷衍生物作为人类的甾体5α-还原酶-II的抑制剂
Cabeza et al.报告称合成和孕烷衍生物系列的人类甾体5α还原酶-II抑制剂活性具有结构多样性和效能。(73)-(79)最好的模型统计分析是采用PLS分析,显示了非常好的交叉验证相关系数Q2 (0.881),非验证相关系数R2数值为(0.893),以及高F-test数值(175.527),伴随着良好的相关性和预测能力。
(39)
三维定量构效关系的空间图显示一坨红点在C-3、C-6和C-17的甾体构架处,说明优良的相互作用的大型取代基存在。同时,在静电势图中,类固醇构架的C-3,C-6,C-16,C-17周围的一坨蓝点显示了电负性取代基的存在,而在C-4和C-16附近的一些红点显示正电性基团存在,这有利于和酶的相互作用。
6-7的不饱和度并没有什么深远的的影响,而在16-17结果化合物具有优良的抑制性活性。图7的分子结构图是通过可视化的空间位阻和静电孕烷衍生物的图设计的。一个QSAR的研究表明,一个庞大的电负性取代基在C-6和C-17和C-4和C-16以及Δ(16,17)与和甾体5α-还原酶-II有良好相互作用关系。
5.4. 6-Azasteroids As Dual Inhibitors of Both Isoforms of Steroidal 5α-Reductase
6-氮杂类甾醇作为甾体5α-还原酶的两个亚型的双重抑制剂
Frye et al.(80)-(82)发布了不同的6 -氮杂甾族(图8)作为甾体5α-还原酶-I和-II的双重抑制剂的合成与生物评价
一个3D-QSAR研究是在选出来的氮杂甾族数据的基础上为两个亚型的甾体5α-还原酶比较6-氮杂甾族产生药效的模型来建立的。(20)还原酶Ⅱ的结构相似性与令人满意的相关性和良好的预测和测试是能力选择的标准。
因为对比模型的发展,甾体5α-还原酶-
I的静电主图,同样的分子被用于甾体5α-还原酶-I和-II的实验集。较少的蓝点在取代基R1和R2被发现。另外,在红点附近,也发现了一些蓝点,表示R3的分支有了选择性和有效的甾体5α-还原酶-I设计的电负性功能。甾体5α-还原酶-II的静电图也显示一些蓝色的点在C-3的甾核存在,确定电负性取代基存在,而红点围绕R1和R2指定为与甾体5α-还原酶-II的正电性取代基存在良性互动。
R3附近的一群红点显示了有效可选的甾体5α-还原酶-II抑制剂活动伴随正电性分支的正电性取代基的存在。甾体5α-还原酶-II的空间主图还显示,存在一群蓝点在“R1”和“R2”在甾体骨架处,说明空间的相互作用的不利于甾体5α-还原酶-II抑制剂的活性。
一簇红点在取代基R3处,显示由于膨胀/分支侧链,有利的空间相互作用,而在附近几个蓝点表示不利的空间相互作用。
伴随-CH3和-CI的R1和R2区域致使复合物拥有增强的5α-还原酶-I抑制剂活动,同时存在微弱的5α-还原酶-II抑制剂活动的降低。
因此,上述替换后显示的完整的分析表明减少甾体5α-还原酶-II同时,观察甾体5α-还原酶-I抑制性被显著提高。总而言之,建议的组合体为设计新的具有强大的,选择性的抑制活性甾体类分子是可以接受的。可见的分子结构的甾体5α还原酶-I和二异构体的空间位阻和静电的图分别是图9和10所示的设计。
图6.目前的3D-QSAR研究中采用不同的孕烷支架作为代表性结构。
图7.为构建有力的人类甾体5α-还原酶-II抑制剂而对孕烷衍生物的静电和空间位阻有要求
图8. 使用6 -氮杂甾族作为目前3D-QSAR 研究的代表性构造支架。
6. CONCLUSIONS 结论
选择甾体5 -还原酶抑制剂是治疗良性前列腺增生症及其他皮肤病的主要干预方法。
对于生产分子结构框架,不同的药效团模型使用各种各样的数据集进行3D-QSAR 研究开发,包括4-氮砸类固醇(非那雄胺类似物),3-羧基类固醇(依立雄胺类似物)、孕烷衍生物和6-氮砸类固醇。两种甾体5α
还原酶亚型之间的相似性已经被通过对比药效突出的3D-QSAR 模型解决。设计新的、有效的甾体核结构时得出的一个组合的结构框架(图11),它与现已提出的需要空间位阻和静电图的3D-QSAR 甾体5α还原酶-II 具有良好的互动性。
我们研究的结果显示,我们所构建的模型是可靠的,可以应用到合理的设计和库筛选中。
比较3D-QSAR 在许多数据集中关于甾体支架探讨甾体类化合物的结构特征时的表现,它能用于修改分子结构以开发选择性抑制剂的两种异构体。
因为研究主要在甾体5α-还原酶的领域,特别是人类甾体5α-还原酶抑制剂的发展,所以它仍然是新兴的,并导致了现在与2D-QSAR 研究结果结合的3D-QSAR 研究,提供了有意义的信息,设计出了在分子识别和目标结合方面更先进的抑制剂,有利于前列腺增生症的治疗。
图9. 为构建有力的人类甾体5α-还原酶-I 抑制剂而对6 -氮杂甾族的静电和空间位阻有要求。
图10. 为构建有力的人甾体5α-还原酶-II抑制剂而对6 -氮杂甾族的静电和空间位阻有要求。图11. 为设计新的对甾体5α-还原酶-II有良好效果的抑制剂的甾体的核的结构框架。
AUTHOR INFORMATION 作者信息Corresponding Author联系人
*E-mail: sureshthareja@https://www.360docs.net/doc/f512790894.html,, sureshthareja@hotmail. com. Phone: +91-9617605869 (M); +91-7752-260027 (O).
Notes注解
作者声明:不可用于商业盈利。
Biography档案
Suresh Thareja博士(生于1983.11.11,印度哈里亚纳邦)目前作为助理教授工作于印度比拉斯普尔市,古鲁加思达斯中央大学,医药科学院。他在MDU大学被授予学士学位(2004),在普纳大学药剂学的药物化学专业获得硕士学位(2007),在昌迪加尔的旁遮普邦大学的药剂化学专业获得博士学位(2011)。
在博士毕业后,他在印度诺华医疗作为科学写手工作。在2012年,他加入了比拉斯普尔市古鲁加思达斯中央大学,作为医药科学院的一员。他的研究领域包括基于计算机的药物设计和实验,以及新型PTP-1B抑制剂和甾体5α还原酶抑制剂的评估。
他在国内和国外的许多同行评价杂志上发表过多篇论文。他也是他所研究的领域的许多国内和国际的奖项的获得者。
ACKNOWLEDGMENTS 感谢
作者非常的感谢Daniel Robinson博士(计算机科学研究组,英国,牛津大学),为他的SOMFA软件,和UIPS的所有分子模型研究小组的成员,旁遮普邦大学,为他们在建立甾类化合物模型时一直的支持,建议和鼓励。
REFERENCES参考文献
(1) Aggarwal, S.; Thareja, S.; Verma, A.; Bhardwaj, T. R.; Kumar, M. Steroids 2010, 75, 109.
(2) Jarman, M.; Smith, H. J.; Nicholls, P. J.; Simons, C. Nat. Prod. Rep. 1998, 15, 495. (3) Banday, A. H.; Shameem, S. A.; Jeelani, S. Steroids 2014, 92, 13.
(4) Shirakawa, T.; Okada, H.; Acharya, B.; Zhang, Z.; Hinata, N.; Wada, Y.; Uji, T.; Kamidono, S.; Gotoh, A. Prostate 2004, 58, 33.
(5) Andersson, S.; Russell, D. W. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 3640.
(6) Silver, R. I.; Wiley, E. L.; Thigpen, A. E.; Guileyardo, J. M.; McConnell, J. D.; Russell, D. W. J. Urol. 1994, 152, 438.
(7) Azzouni, F.; Godoy, A.; Li, Y.; Mohler. J. Adv. Urol. 2012, 2012,1. 18.
(8) Zhu, Y. S.; Sun, G. H. J. Med. Sci. 2005, 25,1.
(9) Uemura, M.; Tamura, K.; Chung, S.; Honma, S.; Okuyama, A.; Nakamura, Y.; Nakagawa, H. Cancer Sci. 2008, 99, 81.
(10) Arena, F. Minerva. Urol. Nefrol. 2008, 60,71.76.
(11) Andersson, S.; Russell, D. W. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 3640.
(12) Sinclair, R.; Patel, M.; Dawson, T. L., Jr.; Yazdabadi, A.; Yip, L.; Perez, A.; Rufaut, N. W. Br. J. Dermatol. 2011, 165, 12.
(13) Metcalf, B. W.; Levy, M. A.; Holt, D. A. Trends Pharmacol. Sci. 1989, 10, 491. Chandigarh (2011). He is the recipient of GATE and ICMR-Senior
(14) Brooks,
J. R.; Berman, D.; Glitzer, M. S.; Gordon, L. R.; Primka, R.
Research Fellowship. After completing his Ph.D. (2011), he worked as L.; Reynolds, G. F.; Rasmusson, G. H. Prostate 1982, 3, 35. (15) Li, J.; Ding, Z.; Wang, Z.; Lu, J. F.; Maity, S. N.; Navone, N. M.; Logothetis, C. J.; Mills, G. B.; Kim, J. PLoS One 2011, 6, e28840.
(16) Baxter, F. O.; Trivic, S.; Lee, I. R. J. Steroid Biochem. Mol. Biol. 2001, 77, 167.
(17) Labrie, F.; Sugimoto, Y.; Luu-The, V.; Simard, J.; Lachance, Y.; Bachvarov, D.; Leblanc, G.; Durocher, F.; Paquet, N. Endocrinology 1992, 131, 1571.
(18) Oliveira, I. O.; Lhullier, C.; Brum, I. S.; Spritzer, P. M. Braz. J. Med. Biol. Res. 2003, 36, 1447.
(19) Salvador, J. A. R.; Pinto, R. M. A.; Silvestre, S. M. J. Steroid Biochem. Mol. Biol. 2013, 137, 199.
(20) Thareja, S.; Rajpoot, T.; Verma, S. K. Steroids 2015, 95, 96.
(21) D. Abell, A.; Brandt, M.; A. Levy, M.; A. Holt,
D. J. Chem. Soc., Perkin Trans. 1 1997, 1663.
(22) Wakeling, A. E.; Furr, B. J. A.; Glen, A. T.; Hughes, L. R. J. Steroid Biochem. 1981, 15, 355.
(23) Kim, S.; Kim, Y. U.; Ma, E. Molecules 2011, 17, 355.
(24) Monda, J. M.; Oesterling, J. E. Mayo Clin. Proc. 1993, 68, 670. 679.
(25) Marks, L. S. Rev. Urol. 2004, 6, 11.
(26) Moss, M. L.; Kuzmic, P.; Stuart, J. D.; Tian,
G.; Peranteau, A. G.; Frye, S. V.; Kadwell, S. H.; Kost, T. A.; Overton, L. K.; Patel, I. R. Biochemistry 1996, 35, 3457.
(27) Sun, J.; Xiang, H.; Yang, L. L.; Chen, J. B. Curr. Med. Chem. 2011, 18, 3576.
(28) Brandt, M.; Greway, A. T.; Holt, D. A.; Metcalf, B. W.; Levy, M. A. J. Steroid. Biochem. Mol. Biol. 1990, 37, 575.
(29) Chen, G. S.; Chang, C. S.; Kan, W. M.; Chang, C. L.; Wang, K. C.; Chern, J. W. J. Med. Chem. 2001, 44, 3759.
(30) Hamza, A.; Wei, N. N.; Zhan, C. G. J. Chem. Inf. Model. 2012, 52, 963.
(31) Berenger, F.; Voet, A.; Lee, X. Y.; Zhang, K. Y. J. Cheminf. 2014, 6, 66.
(32) Butkiewicz, M.; Lowe, E. W., Jr.; Mueller, R.; Mendenhall, J. L.; Teixeira, P. L.; Weaver, C. D.; Meiler, J. Molecules 2013, 18, 735.
(33) Sharma, B. K.; Singh, P.; Kumar, R.; Sharma, S. J. Enzyme Inhib. Med. Chem. 2008, 23, 50.
(34) Hung, C. L.; Chen, C. C. Drug Dev. Res. 2014, 75, 412.
(35) Kumar, R.; Kumar, M. Med. Chem. Res. 2013, 22, 105.
(36) Aggarwal, S.; Thareja, S.; Bhardwaj, T. R.; Kumar, M. Eur. J. Med. Chem. 2010, 45, 476. (37) Aggarwal, S.; Thareja, S.; Bhardwaj, T. R.; Kumar, M. Lett. Drug Des. Discovery 2010, 7, 596.
(38) Thareja, S.; Aggarwal, S.; Bhardwaj, T. R.; Kumar, M. Eur. J. Med. Chem. 2009, 44, 4920.
(39) Aggarwal, S.; Thareja, S.; Bhardwaj, T. R.; Kumar, M. Steroids 2010, 75, 411.
(40) Aggarwal, S.; Thareja, S.; Verma, A.; Bhardwaj, T. R.; Kumar, M. Acta Pol. Pharm. 2011, 68, 447.
(41) Kumar, R.; Malla, P.; Verma, A.; Kumar, M. Med. Chem. Res. 2013, 22, 4568.4580. (42) Aggarwal, S.; Thareja, S.; Bhardwaj, T. R.; Haupenthal, J.; Hartmann, R. W.; Kumar, M. Eur. J. Med. Chem. 2012, 54, 728.
(43) Kurup, A.; Garg, R.; Hansch, C. Chem. Rev. 2000, 100, 909.
(44) Hutter, M. C.; Hartmann, R. W. QSAR Comb. Sci. 2004, 23, 406.
(45) Melo-Filho, C. C.; Braga, R. C.; Andrade, C.
H. Curr. Comput.Aided Drug Des. 2014, 10, 148.
(46) Asakawa, N.; Kobayashi, S.; Goto, J.; Hirayama, N. Int. J. Med.Chem. 2012,9. (47) Asikainen, A. H.; Ruuskanen, J.; Tuppurainen, K. A. Environ. Sci. Technol. 2004, 38, 6724.
(48) Cramer, R. D.; Patterson, D. E.; Bunce, J. D. J. Am. Chem. Soc. 1988, 110, 5959.
(49) Special Issue: Challenges in Virtual Screening. QSAR Comb. Sci. 2006, 12, 1203. (50) Dixon, S. L.; Smondyrev, A. M.; Rao, S. N. Chem. Biol. Drug Des. 2006, 67, 370.
(51) Ghasemi, J. B.; Davoudian, V. J. Chem. 2014, 2014, 10. Open Bioinf. J.
(52) Singh, A.; Singh, R. 2013, 7, 63.
(53) Pahwa, P.; Papreja, M. Acta Pol. Pharm. 2012, 69, 535.
(54) Robinson, D. D.; Winn, P. J.; Lyne, P. D.; Richards, W. G. J. Med. Chem. 1999, 42, 573.
(55) Sun, P.-H.; Yang, Z.-Q.; Li, M.-K.; Chen, W.-M.; Liu, Q.; Yao, X.S. Lett. Drug Des. Discovery 2009, 6, 568.
(56) Baurin, N.; Vangrevelinghe, E.; Allory, L. M. J. Med. Chem. 2000, 43, 1109.
(57) Kansal, N.; Silakari, O.; Ravikumar, M. Lett. Drug Des. Discovery 2008, 5, 437.
(58) Li, M.; Du, L.; Wu, B.; Xia, L. Bioorg. Med. Chem. 2003, 11, 3945.
(59) Korhonen, S. P.; Tuppurainen, K.; Asikainen, A.; Laatikainen, R.; Pera..kyla,M. QSAR Comb. Sci. 2007, 26, 809.
(60) Li, M.-Y.; Fang, H.; Xia, L. Bioorg. Med. Chem. Lett. 2005, 15, 3216.
(61) Li, M.; Xia, L. Chem. Biol. Drug Des. 2007, 70, 461.
(62) Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H. J. J. Comput. Biol. 2008, 15, 737.
(63) Li, S.; Zheng, Y. Int. J. Mol. Sci. 2006, 7, 220.
(64) Thareja, S.; Kokil, G.; Aggarwal, S.; Bhardwaj, T. R.; Kumar, M. Chem. Pharm. Bull. 2010, 58, 526.
(65) Thareja, S.; Aggarwal, S.; Bhardwaj, T. R.; Kumar, M. Eur. J. Med. Chem. 2010, 45, 2537.
(66) Thareja, S.; Aggarwal, S.; Bhardwaj, T. R.; Kumar, M. Med. Chem. 2010, 6, 30.
(67) Thareja, S.; Aggarwal, S.; Bhardwaj, T. R.; Kumar, M. Lett. Drug Des. Discovery 2010, 7, 395.
(68) Goel, H.; Sinha, V. R.; Thareja, S.; Aggarwal, S.; Kumar, M. Int. J. Pharm. 2010, 415, 158. (69) Kulkarni, S. S.; Patel, M. R.; Talele, T. T. Bioorg. Med. Chem. 2008, 16, 3675.
(70) Rasmusson, G. H.; Reynolds, G. F.; Steinberg, N. G.; Walton, E.; Patel, G. F.; Liang, T.; Cascieri, M. A.; Cheung, A. H.; Brooks, J. R.; Berman, C. J. Med. Chem. 1986, 29, 2298. (71) Liang, T.; Cascieri, M. A.; Cheung, A. H.; Reynolds, G. F.; Rasmusson, G. H. Endocrinology 1985, 117, 571.
(72) Holt, D. A.; Levy, M. A.; Oh, H. J.; Erb, J. M.; Heaslip, J. I.; Brandt, M.; Lan-Hargest, H. Y.; Metcalf, B. W. J. Med. Chem. 1990, 33, 943. (73) Cabeza, M.; Zambrano, A.; Heuze, I.; Carrizales, E.; Palacios, A.; Segura, T.; Valencia, N.; Bratoeff, E. Steroids 2009, 74, 793. (74) Ramirez, E.; Cabeza, M.; Bratoeff, E.; Heuze, I.; Perez, V.; Valdez, D.; Ochoa, M.; Teran, N.; Jimenez, G.; Ramirez, T. Chem. Pharm. Bull. 2005, 53, 1515.
(75) Bratoeff, E.; Segura, T.; Recillas, S.; Carrizales, E.; Palacios, A.; Heuze, I.; Cabeza, M. J. Enzyme Inhib. Med. Chem. 2009, 24, 655. (76) Bratoeff, E.; Sainz, T.; Cabeza, M.; Heuze, I.; Recillas, S.; Perez, V.; Rodriguez, C.; Segura, T.; Gonzales, J.; Ramirez, E. J. Steroid Biochem. Mol. Biol. 2007, 107, 48.
(77) Bratoeff, E.; Cabeza, M.; Perez-Ornelas, V.; Recillas, S.; Heuze, I. J. Steroid Biochem. Mol. Biol. 2008, 111, 275.
(78) Perez-Ornelas, V.; Cabeza, M.; Bratoeff, E.; Heuze, I.; Sanchez, M.; Ramirez, E.; Naranjo-Rodriguez, E. Steroids 2005, 70, 217.
(79) Cabeza, M.; Bratoeff, E.; Heuze, I.; Rojas,
A.; Teran, N.; Ochoa, M.; Ramirez-Apan, T.; Ramirez, E.; Perez, V.; Gracia, I. J. Enzyme Inhib. Med. Chem. 2006, 21, 371.
(80) Frye, S. V.; Haffner, C. D.; Maloney, P. R.; Mook, R. A., Jr.; Dorsey, G. F., Jr.; Hiner, R. N.; Cribbs, C. M.; Wheeler, T. N.; Ray, J. A.; Andrews, R. C.; et al. J. Med. Chem. 1994, 37, 2352.
(81) Frye, S. V.; Haffner, C. D.; Maloney, P. R.; Mook, R. A., Jr.; Dorsey, G. F., Jr.; Hiner, R. N.; Batchelor,K. W.; Bramson, H. N.; Stuart, J. D.; Schweiker, S. L.; et al. J. Med. Chem. 1993, 36, 4313.
(82) Frye, S. V.; Haffner, C. D.; Maloney, P. R.; Hiner, R. N.; Dorsey, G. F.; Noe, R. A.; Unwalla, R. J.; Batchelor, K. W.; Bramson, H. N.; Stuart, J.
D.; et al. J. Med. Chem. 1995, 38, 2621.
(83) Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.-
D.; Lee, K.-H.; Tropsha, A. J. Comput.-Aided Mol. Des. 2003, 17, 241.
(84) Sachan, N.; Kadam, S. S.; Kulkarni, V. M. J. Enzyme Inhib. Med. Chem. 2007, 22, 267. (85) Skold, C.; Karlen, A. J. Mol. Graphics Modell. 2007, 26, 145.
(86) SOMFA2 v2.0.0 can be downloaded from http://bellatrix.pcl.ox. https://www.360docs.net/doc/f512790894.html, (2007). 2893 DOI: 10.1021/cr5005953 Chem. Rev. 2015, 115, 2883.2894
HMG-CoA还原酶抑制剂-他汀类药物的作用及机制研究
HMG-CoA还原酶抑制剂-他汀类药物的作用及机制研究 他汀类药物作为 HMG-CoA 还原酶抑制剂 , 能有效降低低密度脂蛋白(LDL) 中胆固醇水平 , 从而大大减少了致命性和非致命性心血管疾病事件的发生。本文就心血管疾病的发病机制 , 以及他汀类药物对心血管疾病的治疗作用等方面进行概述。 心血管疾病又称为循环系统疾病 , 泛指由于高脂血症、血液黏稠、动脉粥样硬化、高血压等所引起的一系列涉及循环系统疾病 , 主要包括冠心病、心绞痛、心肌梗死、高血脂和运动猝死等疾病。 1心血管疾病的发病机制 心血管疾病种类较多 , 大多数都与动脉粥样硬化密切相关。其发病机制可能如下:①高脂血、NO 等因素使血管内壁细胞损伤 , 导致血管壁通透性改变 , 大量脂质透过内皮细胞深入内皮细胞间隙。进而引发内膜增厚、脂质沉积、细胞浸润、中膜平滑肌细胞向管腔转移和增殖等症状。②细胞外基质增生和出现泡沫细胞 , 使动脉壁变成糜粥样结构。 他汀类降脂药为细胞内胆固醇合成限速酶 , 即 3- 羟基 -3- 甲基戊二酰辅酶 A( HMG-CoA) 还原酶的抑制剂 , 是目前临床上应用最广泛的一类降脂药物, 常用的有辛伐他汀、洛伐他汀、普伐他汀、阿托伐他汀、匹伐他汀和氟伐他汀等。 2他汀类药物的结构 他汀类药物的化学结构大致分为三部分:一是与天然底物 HMG-CoA 类似的 3, 5- 二羟基庚酸结构片段 ( 在辛伐他汀和洛伐他汀结构中 , 与天然底物 HMG-CoA 类似的是 3-羟基戊内酯环 , 给药后在体内开环转化为有效的羟基酸 ), 它是他汀类药物的药效团;二是以共价键与 3, 5- 二羟基庚酸相连接的疏水性环状结构 , 它在药物与还原酶的结合中起着重要作用;三是疏水性环状结构上的取代基 , 它们决定药物溶解性和药动学性质。 3他汀类药物的降胆固醇作用 HMG-CoA 还原酶是人体胆固醇合成过程中的关键酶之一 , 他汀类药物具有与其天然底物 HMG-CoA 类似的结构片段 , 与 HMG-CoA 还原酶有着更高的亲和性 , 能够竞争性抑制 HMG-CoA 还原为甲羟戊酸 , 从而减少胆固醇的合成量。上调肝细胞膜上 LDL 受体数目 , 增加的 LDL 受体介导的 LDL 和 IDL( 中间密度脂蛋白 ) 的清除 , 从而使 LDL 和VLDL( 极低密度脂蛋白 ) 的合成下
第八章_非甾体抗炎药
第八章非甾体抗炎药 非甾体抗炎药(Nonsteroidal Antiinflammatory Drugs, NSAIDS)是一类具有抗炎作用和解热、镇痛作用药物。临床上用于治疗胶原组织疾病,例如风湿、类风湿性关节炎,骨关节炎等。此类药物的化学结构与皮质激素类抗炎药物不同,因此被称为非甾体抗炎药。抗炎作用机制与其在体内抑制前列腺素(Prostaglandines, PGs)的生物合成有关。 已经证明前列腺素是一类致热物质,其中前列腺素E2(PGE2)致热作用最强。前列腺素本身致痛作用较弱,但能增强其他致痛物质例如缓激肽、5-羟色胺等的致痛作用,使疼痛加重。另外,前列腺素也是一类炎症介质。非甾体抗炎药通过抑制环氧合酶(Cyclo-oxygenase,COX)阻断前列腺素的生物合成发挥消炎、解热镇痛作用。近年来发现环氧合酶有COX1和COX2两种亚型,COX2是导致炎症反应的酶,因此寻找高选择性的COX2抑制剂可得到更安全的药物。 非甾体抗炎药按化学结构类型分为:水杨酸类、乙酰苯胺类、吡唑酮类、3,5-吡唑烷二酮类、芳基烷酸类、邻氨基苯甲酸类、1,2-苯并噻嗪类等。 一、水杨酸类 阿司匹林(Aspirin)已在临床应用了100多年,为有效的解热镇痛药,用于治疗伤风、感冒、头痛、神经痛、风湿痛及类风湿痛等。近年来经研究发现阿司匹林为不可逆的环氧合酶抑制剂。阿司匹林还能抑制血小板中血栓素 A2(TXA2)的合成,阿司匹林现已用于心血管系统疾病的预防和治疗。长期服用阿司匹林有时可导致胃肠道出血,这是由于抑制了前列腺素的合成,致使胃粘膜失去了前列腺素对它的保护作用,造成胃部血流减少,缺血而引起溃疡。另外阿司匹林及水解产物水杨酸酸性较强对胃粘膜有刺激性,甚至引起胃出血。因此,对阿司匹林进行一系列结构修饰,利用水杨酸分子中的活性功能基羧基将其制成盐、酰胺、酯类以降低羧酸对胃肠道的刺激性。如在临床上应用的有乙酰水杨酸铝(Aluminum acetyl salicylate),乙酰水杨酸赖氨酸盐--赖氨匹林
常用非甾体类抗炎药有哪些
常用非甾体类抗炎药有哪些 来源:寻医问药网 非甾体类抗炎药(NSAIDs)是当今世界各国广泛应用的一类药物,每天全世界约有3千万人使用,每年的处方量达5亿。40%使用者年龄超过40岁,由于非处方药的增加、人口老龄化,而且认识到不仅应用于类风湿关节炎、骨关节炎、其他类型的关节炎;还可治疗与关节炎有关的疾病,以及其他类型的疼痛,并且用于结肠癌、阿尔茨海默病的预防等,因此,非甾体类抗炎药的用量正在逐年增加。 非甾体类抗炎药是治疗类风湿关节炎的首选药物,这一大类抗炎药有百余种之多,我国目前上市的抗炎药物的种类和剂型也不少。有不同结构的药物:如水杨酸类的阿司匹林,吲哚类的消炎痛,苯乙酸类的双氯芬酸钠(扶他林)和丙酸类的布洛芬及芬必得等。有不同半衰期的药物:如短效的布洛芬,中效的萘普生,长效的炎痛喜康等。药物剂型有普通片剂、肠溶片、缓释剂、栓剂、凝胶剂、针剂等。现择其常用的几种介绍于下: (1) 阿司匹林(Asprin) 又称醋柳酸或乙酰水杨酸。水杨酸是1838年从柳木的树皮中提取出来的,1852年化学合成,1890年合成阿司匹林,至今已是百岁寿星。阿司匹林是一种和缓的抗炎、止痛剂。阿司匹林应用剂量,以能充分缓解症状而不引起中毒为宜。有规律地而不是零星地用药效果较好。多数成年人每日3~5克。老年病人一般对大剂量耐受性较差。症状控制后剂量减半。为减少对胃黏膜的刺激作用,可饭后服用,并在睡前或清晨与食物或抗酸剂同时服用。阿司匹林肠溶片不能完全缓解胃炎问题,而且有不易吸收的缺点。 应用阿司匹林可能产生眩晕、恶心、呕吐、耳鸣、视力减退。据报道,70%病例大便隐血出现阳性,引起溃疡病;极少数可引起过敏反应,如哮喘、皮疹、血管神经水肿等。对肝、肾功能不全或溃疡病、凝血酶原缺乏症的病人,应慎用。 (2)贝诺酯为阿司匹林与对乙酰氨基酚的酯化物,可减少阿司匹林的不良反应,具有抗炎、解热、镇痛的作用。本药疗效可靠、适应性强、毒性低,对胃肠道等不良反应少。每次0.5~1克,每日3次。 (3)吲哚美辛(消炎痛,lndometh~in) 消炎、退热、止痛作用较阿司匹林强。剂量每次25毫克,每日2—3次,饭后或餐中服用。少数每周可增加25毫克,直到获得满意效果或每日最大量150毫克。超过该剂量一般不增加药物效果,却能增加不良反应。 吲哚美辛禁用于孕妇、哺乳期妇女、帕金森病的病人;有精神病、癫痫史,以及对其过敏的病人、活动性或复发的胃及十二指肠溃疡病人则相对禁忌;小儿慎用或忌用。副作用:主要出现胃肠道疾病和消化性溃疡、头痛及其他大脑功能障碍。胃肠道疾患包括消化不良、恶心、腹痛、隐匿性出血及消化性溃疡。头痛为时常感觉前额跳动性疼痛,尤以醒后最甚。其他报道的大脑症状有眩晕、头昏目眩、精神错乱、抑郁、昏昏欲睡、幻觉《抽搐和晕厥。还可有角膜后沉着、视力模糊、肝大、血液病(再生障碍性贫血、溶血性贫血、骨髓抑制、粒细胞缺乏症、血小板减少性紫癜、过敏反应(皮疹、哮喘)、听力障碍、水肿(以眼睑多见)、结节性红斑和脱发等。目前吲哚美辛有普通片剂、肠溶片、胶囊、缓释胶囊、栓剂、针剂6种。据我们观察、,在作用与副作用方面,栓剂优于片剂、胶囊,胶囊又优于片剂,针剂虽能肌内注射,起效较快,但毕竟不太方便,不能常用。 (4)氨糖美辛每片含盐酸氨基葡萄糖75毫克,吲哚美辛Z5毫克。氨基葡萄糖是一种海洋生物制剂,是硫酸软骨素的基本成分,能促进黏多糖的合成,提高关节滑液的黏性。本品能改善关节软骨的代谢,有利于关节软骨的修复,具有明显的消炎镇痛作用,且能缓解非甾体类抗炎药对蛋白多糖化合物合成的阻滞作用,从而降低消炎痛原有的毒副作用。每次1—2片,每日2—3次。肾功能不全及孕妇禁用,胃与十二指肠溃疡及小儿慎用。 (5)舒林酸(奇诺力,Sulindac) 在结构上是消炎痛一类吲哚乙酸的衍生物。它以前体—亚砜形式服用,然后在体内代谢为活性的硫化代谢产物和无活性的砜代谢产物。活性的代谢产物具有可逆的抑制环氧化酶的作用,减少致炎的前列腺素的合成。因为活性的硫化代谢产物在到达肾脏前已变为无活性的砜,或者在肾脏内被氧化酶转变为无活性代谢物,因此对肾脏影响较其他非甾体类抗炎药为小。另外,与其他非甾体类抗炎药不同:本品抑制血小板聚集作用很小,延长出血时间的作用也较阿司匹林为小。本晶对血压控制的影响,也较其他非甾体类抗炎药小。尤其适用于老年病人。每次200毫克,每日2次。
第五章 非甾体抗炎药
第五章非甾体抗炎药 大纲要求: 掌握:解热镇痛药、非甾体抗炎药的结构类型。 掌握:代表药物阿司匹林(典、基)、对乙酰氨基酚(典、基)、吲哚美辛(典、基)、双氯芬酸钠(碘、基)、布洛芬(典、基)的化学名、结构、理化性质和用途。 掌握:贝诺酯(典)、安乃近(典)、萘普生(典、基)的结构、理化性质及用途。 熟悉:萘丁美酮(基)、芬布芬(典)、舒林酸(典、基)、酮洛芬(典)、吡罗昔康(典)、美洛昔康(基)、别嘌醇(典、基)、丙磺舒(典、基)和秋水仙碱(典、基)的结构、作用特点和用途。 了解:解热镇痛药、非甾体抗炎药和抗痛风药的发展和现状。 一、A型题(最佳选择题) 1.以下药物中,哪个药物对COX-2的抑制活性比COX-1的抑制活性强 A.吡罗昔康B.布洛芬C.吲哚美辛D、美洛昔康E、酮洛芬 2.以下药物中,哪个药物以右旋体供药 A.萘普生B.布洛芬C.萘丁美酮D.安乃近E.吡罗昔康 3.下列药物中,具有1,2一苯并噻嗪结构的药物是 A、吡罗昔康B.吲哚美辛C.萘普生D.芬布芬E.非诺洛芬 4.具有下列化学结构的药物是: (以下为化学结构) A、阿司匹林 B、安乃近C.吲哚美辛D.对乙酰氨基酚E.贝诺酯 5、具有下列化学结构的药物是 (以下为化学结构) A、芬布芬 B、萘丁美酮C.吲哚美辛D.对乙酰氨基酚E.安乃近 6.下列药物中哪个药物不溶于碳酸氢钠溶液 A、布洛芬B对乙酰氨基酚C.双氯芬酸D.萘普生E.酮洛芬 7.下列药物中,哪个药物结构中不含有羧基却具有酸性 A.阿司匹林B、吡罗昔康C.布洛芬D.吲哚美辛E.舒林酸 8.非甾体抗炎药的化学结构类型可分为 A.水杨酸类、苯胺类、吡唑酮类 B.3,5一吡唑烷二酮类、芳基烷酸类、芬那酸类、l,2一本并噻嗪类及其他类 C.吲哚乙酸类、芳基烷酸类、水杨酸类 D.吡唑酮类、芳基烷酸类、吲哚乙酸类 E.3,5一吡唑烷二酮类、水杨酸类、l,2一苯并噻嗪类 9、下列哪个药物可以抑制青霉素在肾小管的分泌,增加其血药浓度,延长作用时间 (以下为化学结构式) 10.下列药物中,哪个药物可溶于水 A、吲哚美辛B.吡罗昔康C.安乃近D.布洛芬E、贝诺酯 11.非甾体抗炎药物的作用机制是 A.二氢叶酸还原酶抑制剂B.二氢叶酸合成酶抑制剂C.β一内酰胺酶抑制剂 D.花生四烯酸环氧化酶抑制剂E.粘肽转肽酶抑制剂 12.下列化学结构中哪个是别嘌醇 (以下为化学结构式) 13.药典中采用下列哪种方法检查阿司匹林中的水杨酸杂质 A.检查水溶液的酸性B.检查碳酸钠中的不溶物C.与高铁盐溶液呈色
常用的五大类降脂药
For personal use only in study and research; not for commercial use 常用的五大类降脂药高脂血症根据发生异常改变的血脂成分的不同,可分为以下四 种:1.单纯性高胆固醇血症(正常人的血总胆固醇应低于5.2mmol/L,如超过 5.7mmol/L,即可诊断为高胆固醇血症)。2.单纯性高甘油三酯血症(指血甘油三酯 超过1.7mmol/L)。3.混合型高脂血症(指既有血浆胆固醇水平升高,又有血浆甘油 三酯水平升高)。4.低高密度脂蛋白血症(高密度脂蛋白小于40mg/dl)。目前 ,在临床上常用的降脂药物有许多,归纳起来大体上可分为五大类。 1 他汀类 三甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,即胆固醇生物合成酶抑制剂(他 汀类药物),是细胞内胆固醇合成限速酶,即HMG-CoA还原酶的抑制剂,为目前临床 上应用最广泛的一类调脂药物。由于这类药物的英文名称均含有“statin”,故常
简 称为他汀类。现已有5种他汀类药物可供临床选用:(1)洛伐他汀(lovastatin ),常见药物有美降之、罗华宁、洛特、洛之特等,血脂康的主要成分也是洛伐他汀 。(2)辛伐他汀(simvastatin),常见药物为舒降之、理舒达、京必舒新、泽之浩 、苏之、辛可等。(3)普伐他汀(pravastatin),常用药有普拉固、美百乐镇。(4)氟伐他汀(fluvastatin),常见药有来适可。(5)阿托伐他汀(atorvastatin ),常见药为立普妥、阿乐。他汀类药物是目前治疗高胆固醇血症的主要药物。 该类药物最常见的不良反应主要是轻度胃肠反应、头痛。与其他降脂药物合用时可能 出现肌肉毒性。 2 贝特类贝特类药物的主要适应症为:高甘油三酯血症或
第六章 解热镇痛药及非甾体抗炎药
第三章解热镇痛药及非甾体抗炎药 解热镇痛药是一类能使发热病人的体温降至正常,并能缓解疼痛的药物。其中除乙酰苯胺类外,大多具有抗炎作用。非甾体抗炎药也兼有退热止痛作用,但在临床应用上有所侧重,它主要用于抗炎抗风湿,有的还兼有排尿酸抗痛风作用。 背景知识 炎症:具有血管系统的活体组织对损伤因子所发生的防御反应为炎症。炎症,就是平时人们所说的“发炎”,是机体对于刺激的一种防御反应,表现为红、肿、热、痛和功能障碍。 从炎症的主要的组织变化可分类如下: 1)变质性炎症2)增生性炎症 3)渗出性炎症(浆液性炎症、纤维素性炎症、化脓性炎症、出血性炎症、坏死性炎症等)4)特异性炎症(结核、梅毒、麻疯、淋巴肉芽肿等) 1、抗炎药有两大类:(1)一类是甾体抗炎药,即肾上腺皮质所分泌的糖皮质激素氢化可的松及其人工合成的衍生物,如醋酸氢化可的松、醋酸地塞米松等。(2)另一类是非甾体抗炎药,即医疗实践中所指的解热镇痛抗炎药等,如阿司匹林等本章所涉及药物。 2、非甾体抗炎药:是一类不含有甾体结构的抗炎药,该类药物具有抗炎、抗风湿、止痛、退热和抗凝血等作用。非甾体类抗炎药是治疗类风湿关节炎的首选药物,此外还用于多种发热和各种疼痛症状的缓解。每天全世界约有3千万人使用,每年的处方量达5亿。40%使用者年龄超过40岁。近年研究表明,在产生痛刺激的局部病灶内,前列腺素PG的合成和释放均有增加。 3、前列腺素(PG) 存在于动物和人体中的一类不饱和脂肪酸组成的具有多种生理作用的活性物质。最早发现存在于人的精液中,当时以为这一物质是由前列腺释放的,因而定名为前列腺素。现已证明精液中的前列腺素主要来自精囊,除之全身许多组织细胞都能产生前列腺素。PG是产生炎症的介质,PGE2有很强的微血管扩张作用和致痛增敏作用,使炎症局部出现红、肿、痛、热等一系列反应。 4、作用机制 解热镇痛药和非甾体抗炎药的作用机制相同:通过抑制PG合成酶,使PG的合成和释放减少,达到解热镇痛效果。
六章 芳酸类非甾体抗炎药物的分析习题及答案
六章芳酸类非甾体抗炎药物的分析 一、基本要求 1.掌握:芳酸类非甾体抗炎药物的结构和性质;主要芳酸类药物的鉴别、检查和含量测定的原理与特点。 2.熟悉:主要芳酸类非甾体抗炎药物杂质的结构、危害、检查方法与含量限度。 3.了解:影响芳酸类非甾体抗炎药物稳定性的主要因素,体内样品与临床监测。 二、基本内容 非甾体抗炎药( Non-steroidal Anti-inflammatory Drugs,NSAIDs)是一类不含有甾体骨架的抗炎药,是目前临床使用最多的药物种类之一。本类药物具有不同的化学结构,但多数具有芳酸基本结构,即芳基取代羧酸结构。根据芳基在羧酸的取代位置及芳基上的取代基的不同,芳酸类药物可分为水杨酸(邻羟基苯甲酸)、邻氨基苯甲酸、邻氨基苯乙酸、芳基丙酸、吲哚乙酸及苯并噻嗪甲酸等六类。尼美舒利和对乙酰氨基酚在结构上不属于芳酸类,但作为较常用的非甾体抗炎药将在本章一并介绍。 本类药物的结构特点为同时具有游离羧基和苯环,其酸性特征可作为原料药的含量测定基础,即在中性乙醇或其他水溶性有机溶剂中,用氢氧化钠滴定液直接滴定;苯环的紫外光吸收特性常被用于本类药物的鉴别、定量检查及部分制剂的含量测定。本类药物的酯类易子水解的特性决定了其特殊杂质检查的项目与方法,如阿司匹林中游离水杨酸的检查,ChP 曾采用三价铁比色法检查。但由于在供试品溶液制备过程中阿司匹林的继续水解使检查结果不稳定。所以ChP2010采用1%冰醋酸甲醇溶液制备供试品溶液,以增加阿司匹林的稳定性,同时采用高效液相色谱法( HPLC)检查,以提高检查结果的可靠性。 基于药物结构中游离羧基的酸性和芳环的紫外吸收特性,本类药物原料药的含量测定主要采用酸碱滴定法,制剂的定量检查,如溶出度(释放度)、含量均匀度等主要采用紫外一可见分光光度法,而制剂的含量测定则采用紫外一可见分光光度法和高效液相色谱法。 本类药物的分析方法见表6-1。
Triapine_核糖核苷酸还原酶(ribonucleotide reductase)抑制剂_236392-56-6_Apexbio
核糖核苷酸还原酶(ribonucleotide reductase 客户使用Apexbio产品发表的文献 质量控制 质量控制和MSDS COA (Certificate Of Analysis) HPLC NMR (Nuclear Magnetic Resonance) MSDS (Material Safety Data Sheet) SDF [(E)-(3-aminopyridin-2-yl)methylideneamino]thiourea C1=CC(=C(N=C1)C=NNC(=S)N)N 分子量
溶解性Soluble in DMSO > 10 mM储存条件Store at -20° C 一般建议为了使其更好的溶解,请用37℃加热试管并在超声波水浴中震动片刻。储液可以在零下20℃中保存数月。 运输条件试用装:蓝冰运输。 其他可选规格:常温运输或根据您的要求用蓝冰运输。 生物活性 描述Triapine是一种有效的核糖核苷酸还原酶抑制剂。 靶点ribonucleotide reduct ase IC50 产品描述 Triapine是一种有效的核糖核苷酸还原酶(ribonucleotide reductase)抑制剂,在各种肿瘤细胞系中均具有抑制效应,IC50值为1.6μM [1]。 据报道,Triapine抑制核糖核苷酸还原酶活性。Triapine通过抑制DNA合成和修复,展示了抗肿瘤活性。在小鼠M109肺癌和人A2780卵巢癌异种移植裸鼠中,Triapine抑制肿瘤生长。在L1210白血病细胞中,Triapine在很宽的剂量范围内均可以起作用[1,2]。 参考文献: [1]. Jennifer J. Knox, Sebastien J. Hotte, Christian Kollmannsberger, Eric Winquist, Bryn Fisher, Elizabeth A. Eisenhauer .Phase II study of Triapine? in patients with metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161). nvestigational New Drugs .October 2007, Volume 25, Issue 5, pp 471-477 [2]Finch RA1, Liu M, Grill SP, Rose WC, Loomis R, Vasquez KM, Cheng Y, Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem Pharmacol. 2000 Apr 15;59(8):983-91
醛糖还原酶抑制剂
和力更强,连接更紧密,而且不易水解,作用持续时间更长。被期待用于广泛糖尿病并发症如神经疾病、白内障、视网膜及肾疾病的治疗,目前处于三期临床试验中。 N HN Br F OO O O Ranirestat F ONH HN O OO NH2 NNH HN O OF Br FOO Fidarestat Minalrestat F ONH HN OOSorbinil 海因类 海因类ARIS构效关系研究表明(以fidarestat为例),其结构中的苯并二氢毗喃环可与ALR2多肽链上的氨基酸残基Trp20、Trp111、Phe122和Trp219形成疏水键,同时1- 位O通过水分子与Ala299和Leu300主链上的N形成间接氢键。螺乙内酞脉环上的两个羰基通过亲水作用与酶形成氢键,2’-位羰基与Tyr48的O形成一氢键,5’-位羰基与Trp111的N形成另一氢键。1’位的N与His110的N也适合形成一个氢键,同时氨甲酰基上的O可与ALR2主链Leu300的N形成氢键,这是fidarestat具有较强亲和力的原因。Fidarestat与ALR2主要氨基酸残基作用情况见图1。 O NH N OO O NH2 HNH N
COOH N NH N OH NH HHHTrp219 Leu300Trp111 His110 Tyr48 Phe122 Trp20 图1 fidarestat与ALR2活性位点的主要氨基酸残基相互作用示意图 2. 羧酸类 在海因类ARIs研究的基础上,人们合成了一系列带有环状羟基乙酸结构和色原环骨架的化合物。该类化合物在体内外对ALR2 有较强的抑制活性,而且没有索比尼尔类似的过敏反应,其代表性化合物有依帕司他(epalrestat)、托瑞司他(tolrestat)、苯并噻嗪乙酸衍生物(SG-210)[9]和唑泊司他(zopolrestat) 。epalrestat已经通过三期临床试验, 20世纪90年代在日本和欧洲上市,但至今仍没有获得美国FDA的许可。依帕司他可以有效预防并且改善糖尿病并发的末梢神经障碍、震动感觉异常和心搏异常等症状,其渗透性和生物利用度都相当高[,轻微的副作用发生率很低。托瑞司他在1989 年曾以Alredase 为商品名在爱尔兰上市, 用于治疗糖尿病继发的周围感觉性神经疾病。但在后续的治疗糖尿病并发的神经病变的大规模随机双盲临床试验中, 托瑞司他因未能表现出足够的疗效, 未能通过FDA。苯并噻嗪乙酸衍生物(SG-210)对于部分提纯的ALR2,有非常高的活性,吸收快、半衰期长、生物利用度
非甾体抗炎药综述
一.简介 非甾体抗炎药( no n- steroidal ant i- inf lammatory dr ug s, NSAIDs) 是指具有解热、镇痛和消炎作用而非类固醇结构的药物。临床应用极为广泛, 是仅次于抗感染药的第二大类药物1。非甾体抗炎药是急、慢性风湿性疾病的非类固醇一线治疗药物, 具有抗炎、止痛和解热等作用, 主要用于炎症免疫性疾病的对症治疗, 能有效缓解肌肉、关节及炎症免疫性疾病的局部疼痛、肿胀等, 广泛用于腰背痛、牙痛、痛经、急性痛风、外伤或手术后疼痛、癌痛等的治疗, 且无成瘾性和依赖性的特点。据不完全统计, 全世界大约有1亿多人在服用NSA ID s, 其中有一半以上是老年患者。每天约有3 0 0 0 万关节炎患者服用NSAIDs,在我国最保守估计每年至少有500 万OA患者和4 2 0 万R A 患者在服用N S A I D s 。在中国由于各种原因引起的急慢性疼痛的患者约占门诊总人数的1/ 5~ 1/ 4, 因此, 可以说N SA ID s 是临床医师特别是骨科大夫应用较多的药物之一2。随着此类药物的研究进展, 其临床使用范围在不断扩展。 二.发展简史 以阿司匹林为代表的N S A I D s ,具有神奇的、源远流长的历史。追溯到公元前约460 年至377 年希波克拉底曾经使用柳树皮来治疗骨骼肌肉疼痛;1 7 6 3 年的英国传教士爱德蒙特·斯通(E d m a n dS t o n e )第一次比较科学的描述将柳树叶煎液作为一种抗炎药;1828 年德国慕尼黑药学教授约翰·布赫勒(Johann Buchner)提取出柳树皮中的有效成分水杨苷,次年汉立·里劳西(Henri Leroux)获得其结晶;水杨酸则是意大利化学家雷非·皮立亚(Raffaele Piria)首次从水杨苷中获得,1859年德国化学家赫尔曼·柯比(Hermann Kolbe)完成了鉴定及合成其化学结构的工作,1 8 7 4 年水杨酸开始生产;鉴于水杨酸的胃肠道刺激性和不适的口感,1 8 9 7 年德国拜耳公司的化学家霍夫曼(H o f f m a n n )成功合成了乙酰水杨酸;随后拜耳公司的首席药理学家海里希·狄里舍(H e i n r i c hDresser)通过自身实验和随后的动物实验证明乙酰水杨酸具有良好的抗炎和镇痛作用,并于1899 年注册了商品名为阿司匹林(Aspirin)。此后的100 多年来,阿司匹林深受医生和患者的青睐,作为NSAIDs的原形药并成为药物史上的一颗“常青树”3。 1898 年, 由德国拜尔药厂首先合成的阿司匹林是最早用于风湿热及关节炎的治疗药物。由于其在大剂量时才能发挥消炎止痛作用并伴随明显的胃肠道副作用, 逐渐被新上市的NSAID 所取代。1952 年, 保泰松( 苯丁唑酮) 问世, 为第一个被命名的非甾体抗炎药。因其强大的抗炎镇痛效应而广泛用于风湿病的治疗长达30 多年。至20 世纪80 年代, 因相关的不良反应, 如粒细胞和血小板减少, 甚至再生障碍性贫血等不断出现而逐渐被限制使用或禁用。1963 年, 吲哚乙酸类NSAID 的代表药物消炎痛( 吲哚美辛) 上市, 虽然其抗炎、镇痛和解热作用较强, 但因胃肠道、肝脏和肾脏等毒副作用仍然很严重而逐渐少用。1969 年, 1姜爱霞.非甾体抗炎药的研究[ J].潍坊学院学报.2010,10(6):96-98 2刘红,李国珍,葛泉丽.非甾体抗炎药的作用机制及进展[J ].实用医技杂志.2003,10(4):401-402 3李梦涛,曾小峰.非甾体类抗炎药的过去、现在与将来[J ].继续医学教育.2006,20(28):24-29
常用的五大类降脂药
常用的五大类降脂药 高脂血症根据发生异常改变的血脂成分的不同,可分为以下四种:1.单纯性高胆固醇血症(正常人的血总胆固醇应低于5.2mmol/L,如超过5.7mmol/L,即可诊断为高胆固醇血症)。2.单纯性高甘油三酯血症(指血甘油三酯超过1.7mmol/L)。 3.混合型高脂血症(指既有血浆胆固醇水平升高,又有血浆甘油三酯水平升高)。 4.低高密度脂蛋白血症(高密度脂蛋白小于40mg/dl)。 目前,在临床上常用的降脂药物有许多,归纳起来大体上可分为五大类。 1 他汀类 三甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,即胆固醇生物合成酶抑制剂(他汀类药物),是细胞内胆固醇合成限速酶,即HMG-CoA还原酶的抑制剂,为目前临床上应用最广泛的一类调脂药物。由于这类药物的英文名称均含有“statin”,故常简称为他汀类。 现已有5种他汀类药物可供临床选用:(1)洛伐他汀(lovastatin),常见药物有美降之、罗华宁、洛特、洛之特等,血脂康的主要成分也是洛伐他汀。(2)辛伐他汀(simvastatin),常见药物为舒降之、理舒达、京必舒新、泽之浩、苏之、辛可等。(3)普伐他汀(pravastatin),常用药有普拉固、美百乐镇。(4)氟伐他汀(fluvastatin),常见药有来适可。(5)阿托伐他汀(atorvastatin),常见药为立普妥、阿乐。 他汀类药物是目前治疗高胆固醇血症的主要药物。该类药物最常见的不良反应主要是轻度胃肠反应、头痛。与其他降脂药物合用时可能出现肌肉毒性。 2 贝特类 贝特类药物的主要适应症为:高甘油三酯血症或以甘油三酯升高为主的混合型高脂血症。目前临床应用的贝特类药物,主要有环丙贝特、苯扎贝特、非诺贝特及吉非贝齐。据临床实践,这些药物可有效降低甘油三酯22%~43%,而降低TC 仅为6%~15%,且有不同程度升高高密度脂蛋白的作用。该药常见的不良反应为胃肠反应、恶心、腹泻,严重者可导致肝损害。 3 烟酸类 烟酸类药物属B族维生素,当用量超过其作为维生素作用的剂量时,可有明显的降脂作用。该类药物的适用范围较广,可用于除纯合子型家族性高胆固醇血症,及I型高脂蛋白血症以外的任何类型高脂血症。但是,该药的速释制剂不良反应大,我们一般不单独应用。对于烟酸的降脂作用机制,目前医学界尚不十分明确。缓释制剂大大减少,主要为颜面潮红。 4 胆酸螯合剂 这类药物也称为胆酸隔置剂。有考来烯胺(Cholestyramine),常用药物有降胆宁。该药常见的不良反应为胃肠反应、恶心、便秘或腹泻,肠梗阻或头痛等。
第五章第六节:解热镇痛药及非甾体抗炎药(答案)
第五章:解热镇痛药及非甾体抗炎药(自考复习) 一、单 项选择题 ( 在每小题的五个备选答案中,选出一个正确答案,并将正确答 案的序号 1、在阿司匹林合成中产生的可引起过敏反应的副产物是( A )。 A. 乙酰水杨酸酐 B.吲哚 C. 苯酚 D. 水杨酸苯酯 E.乙酰水杨酸苯酯 2、吡唑酮类药物具有的药理作用是( B )。 A. 镇静催眠 C. 抗菌 E.降低血压 4、双氯芬酸钠属于哪类药物 ( B ) B. 阿司匹林 D. 对乙酰氨基酚 8、在阿司匹林合成中产生的可引起过敏反应的副产物是 B.解热镇痛抗炎 D. 抗病毒 3、扑热息痛药物中应检查 ( B ) 杂质的含量。 A. 对乙酰氨基酚 D. 对氨基苯甲酸 B.对氨基酚 C.水杨酸 E.醋酸 A. 邻氨基苯甲酸类 B.吲哚乙酸类 C.芳基烷酸类 D. 吡唑烷酮类 E.苯胺类 5、下列抗炎药与其结构类型不相对应的是 ( D )。 A. 阿司匹林——水杨酸类 B. 吲哚美辛一一杂环芳基乙酸类 C. 萘普生——杂环芳基丙 酸类 D. 布诺芬——吡唑酮类 6、布洛芬是 ( A )。 A. 非甾体抗炎药 C. 心血管药 B. 中枢兴奋药 D. 镇痛药 7、具有手性碳原子,临床上用其( + )异构体的药物是 ( C )。 A. 安乃近 C.萘普生
A. 乙酰水杨酸酐 C. 苯酚 E. 乙酰水杨酸苯酯 9、非甾体消炎药是通过抑制花生四烯酸 作用的( A )。 C. 去甲肾上腺素 D.色胺 12、下列药物中,没有镇痛作用的是 (C )。 A. 阿司匹林 B.海洛因 C. 扑热息痛 D.苯妥英钠 13、下列镇痛消炎药中,具有芳基丙酸结构的是 (D )。 A. 阿司匹林 C. 保泰松 D.布洛芬 14、力口 FeCb 立即显稳定红色的是( D ) A. 氨甲丙二酯 B .卡马西平 C. 苯巴比妥 .安疋 B.吲哚 D.水杨酸苯酯 1),阻断(2)的合成,而显示消炎、解热、镇痛 A. ( 1)环氧合酶(2)前列腺素 C. ( 1)环氧合酶(2)白三烯 10. 下列 药物中没有消炎作用的是 (B A. 阿司匹林 C. 萘普生 11、 抗抑郁药盐酸氟西汀是哪种生物胺的[ B. ( 1)酯氧酶(2)前列腺素 D. ( 1)酯氧酶(2)白三烯 ) 。 B. 扑热息痛 D. 吲哚美辛 聂取的抑制剂 ?( B ) .5-羟色胺 B.吲哚美辛 D.氯丙嗪 15、 扑热息痛药物中应检查 ______ A. 对乙酰氨基酚 C. 水杨酸 E. 醋酸 16、 具有手性碳原子,临床上用其( 杂质的含量。(B ) B. 对氨基酚 D. 对氨基苯甲 酸 + )异构体的药物是(C )
HMG-CoA还原酶抑制剂研究进展
HMG-CoA还原酶抑制剂的研究进展 关键词:HMG-CoA还原酶抑制剂他汀类构效关系降血脂药 HMG-CoA还原酶即3-羟基-3-甲基戊二肽辅酶A(3-Hydroxy-s-Methylglutary-Coenzyme),简称HMGR,是胆固醇生物合成的限速酶。它是降血脂药物设计的重要靶标,抑制该酶的活性可以有效地降低血浆总胆固醇(Choleterol.CH)水平,从而降低罹患心脑血管疾病的几率[1]。HMG-CoA还原酶抑制剂(HMGRI)是一类新型降血脂药物,对HMG-CoA有抑制作用。以他汀类药物为代表,具有选择性好、疗效高、副作用少,是目前临床应用最广、疗效最好、深受广大医生和患者好评的降血脂药。 1.HMG-CoA还原酶抑制剂的分类 目前已上市的HMG-CoA还原酶抑制剂(他汀类)有洛伐他汀(Lovastatin)、辛伐他汀(Simvastatin)、普伐他汀(Pravastatin)、阿托伐他汀(Atovastatin)、氟伐他汀(Fluvastatin)、长效氟伐他汀缓释片、西立伐他汀(Cerivastatin),由于其严重的横纹肌溶解副作用已从市场撤销。最新上市的有罗苏伐他汀(Rosuvastatin)、匹伐他汀(Pitavastatin)。其中,匹伐他汀是一个潜在的“超级他汀”,低剂量降LDL-C疗效与十倍剂量的阿托伐他汀相似,且对糖尿病合并高胆固醇血症的患者更为有效[2]。 依据他汀类药物的来源的化学结构[3],将来源于微生物培养基、培养液的美伐他汀及其衍生物的洛伐他汀、辛伐他汀和普伐他汀作为第1代他汀类药物,将人工合成的含氮杂环和氟苯环的氟伐他汀、阿托伐他汀视为第2代他汀类药物。 2.HMG-CoA还原酶抑制剂的作用机制[4] 人体胆固醇来源于体内的生化合成和从饮食中摄入,前者约占人体胆固醇的70%—80%。所以降低胆固醇的方法除了注意饮食外,更重要的是抑制其在体内的合成。由于HMG-CoARI与HMG-CoA还原酶底物结构相似,可以竞争结合该酶的活性中心,并抑制其活性,而阻止细胞内胆固醇的合成,从而降低血降胆固醇浓度。 在血浆中低密度脂蛋白(LDL)水平过高时,巨噬细胞等由于摄入了经氧化变性的LDL,变成泡沫细胞,这是动脉硬化的原因,所以为了防止动脉硬化,希望降低血中的LDL、IDL (中密度脂蛋白)和VLDL(极低密度脂蛋白)的水平,增加HDL(高密度脂蛋白)的水平。HMG-CoARI能降低肝细胞内胆固醇的浓度,使血中除去更多的LDL,最终使LDL减少。而LDL 的减少受LDL受体的控制,其受体的浓度又受细胞内胆固醇浓度的调节,所以人肝细胞内胆固醇浓度降低则刺激LDL受体的合成,促使LDL进入肝细胞从而降低血中LDL水平。 3.他汀类药物的结构类型[5] 洛伐他汀以内酯型结构应用,本身无活性,口服后在肝脏内易水解出有活性的β羟酸发挥作用。噻伐他汀为人工半合成的强效HMGR抑制剂,它的化学结构和药理特性与洛伐他汀相似,属前体药物与HMGR的亲和力是洛伐他汀的2倍,所以抑制体内胆固醇的合成效果略强于洛伐他汀。普伐他汀是以美伐他汀为原料经真菌进行微生物转化而制得的HMGR 抑制剂,其活性高于M,但低于L和P。[4] 4.他汀类药物的构效关系[6] HMG-CoA还原酶抑制剂的基本结构见图2,其构效关系分为母环、连接链和侧链3个部分讨论。
甾体5α-还原酶抑制剂讲义
Steroidal 5α-Reductase Inhibitors: A Comparative 3D-QSAR Study Review 甾体5α-还原酶抑制剂:一项类比性的三维定量构效关系(3D-QSAR)研究报告Suresh Thareja* (作者姓名) School of Pharmaceutical Sciences, Guru Ghasidas Central University, Bilaspur, Chhattisgarh 495 009, India 医药科学院,古鲁加思达斯中央大学,比拉斯普尔市,恰蒂斯加尔邦495009,印度 CONTENTS 目录 1. Introduction简介2883 2. Historical Aspect of Published 2D-QSAR Studies 已发布的二维定量构效关系(2D-QSAR)的历史概况2884 3. Current Aspect of 3D-QSAR Studies 三维定量构效关系研究的目前情况2885 4. 3D-QSAR Methodology Employed for Steroidal 5α-Reductase Inhibitors 甾体5α-还原酶抑制剂的三位定量构效关系的研究方法论2885 4.1. Selection of Data Sets and Inhibitory Activities 数据集和抑制活性数据精选2885 4.2. Selection of Training and Test Sets 实验和练习册精选2885 4.3. Molecular Modeling and Alignment 分子建模和校正2886 4.4. 3D-QSAR Models 三维定量构效关系模型2886 4.5. Statistical Analyses 统计学分析2886 5. Results and Discussion of 3D-QSAR Studies on Steroidal 5α-Reductase Inhibitors 甾体5α-还原酶抑制剂的三维定量构效关系研究的结论和研讨2886 5.1. Finasteride Analogues (4-Azasteroids) As Inhibitors of Steroidal 5α-Reductase-II 非那雄胺类似物(4-氮杂类甾醇)作为甾体5α-还原酶-II的抑制剂2886 5.2. Epristeride Analogues (3-Carboxysteroids) As Inhibitors of Steroidal 5α-Reductase-II依立雄胺类似物(3-羧基甾体化合物)作为甾体 5α-还原酶-II的抑制剂2888 5.3. Pregnane Derivatives As Inhibitors of Human Steroidal 5α-Reductase-II孕烷衍生物作为人类的甾体5α-还原酶-II的抑制剂2888 5.4. 6-Azasteroids As Dual Inhibitors of Both Isoforms of Steroidal 5α-Reductase 6-氮杂类甾醇作为甾体5α-还原酶-II的两个亚型的双重抑制剂2888 6. Conclusions结论2890 Author Information 作者信息2892 Corresponding Author 联系人2892 Notes 注解2892 Biography 档案2892 Acknowledgments 特别感谢2892 References 参考文献2892 1. INTRODUCTION 简介 良性前列腺增生(BPH)是由于其机制增生和前列腺的腺体元素造成的非癌前列腺增长。其结果是近端尿道梗阻,从而导致了尿流紊乱①。这是由雄激素双氢睾酮(DHT)的增强水平导致的,这种激素在前列腺生长中起到了重要作用。 甾体5α-还原酶(EC 1.3.99.5)是一种细胞核核膜结合酶,它能在NADPH的辅助下将内源性睾酮(T)转化为双氢睾酮。②DHT在这些组织中刺激细胞增长,因此造成了中老年人体中的前列腺迅速肥大。我们针对使用5α-还原酶抑制从T到DHT的转换过程所提出的化学机理,涉及到了酶和NADPH之间的二元复合物的形成。紧接着的是以T为基层的三元复合物的形成。③甾体5α-还原酶在提升DHT含量的许多情境和恶疾中都扮演着重要角色,包括前列腺增生症,前列腺癌,多毛症,痤疮和男性秃顶。④
第九章非甾体抗炎药
第九章非甾体抗炎药 第一节概论 一、炎症及NSAID S 非甾体抗炎药(NSAID S)是一类临床广泛应用的药物,也是全球用量最大的一类药物,能有效地治疗一些自身免疫性疾病,如风湿性和类风湿性关节炎、骨关节炎、红斑狼疮及强直性脊椎炎等。对感染性炎症也有一定的疗效。与甾体抗炎药相比,NSAID S具有安全性好、毒副反应较小等优点。大多数NSAID S不仅具有抗炎作用,而且兼有解热镇痛之功效,而通常所称的解热镇痛药也有一定的抗炎作用,因此将其包含在NSAID范围之内。治疗急性痛风药与NSAIDS密切相关,故本章对抗痛风药也一并加以介绍。 对于一个药物而言,抗炎、解热、镇痛这三方面的作用并非完全平行。如乙酰水杨酸、荼普生、希洛芬等均具有抗炎、解热、镇痛这三个方面的作用,但每种药物对这三个方面的作用强度是不同的;保泰松、羟布宗仅有抗炎、解热作用镇痛作用微弱;吡罗西康具有抗炎镇痛作用,而无解热作用;乙酰苯胺类如乙酰氨基酚仅有解热、镇痛作用,无抗炎作用。 炎症是对任何刺激的正常和重要反应。是一种很重要的自身防御机制,有害刺激物可威胁患者并可能对患者造成局部刺激。如发热、红肿和疼痛,炎症的顺序可简要归纳如下:(1)初期损伤可引起释放炎症介质 (2)扩张血管 (3)增加血管通透性和渗出液 (4)白细胞渗出、白细胞趋化性和吞噬作用 (5)结缔组织细胞的增生 炎症和关节炎患者的病因近年来引起人们的广泛关注,到目前为止,现有的药物只能对症治疗,可以减轻症状,尚不能治愈。 解热镇痛药物作用于外周神经,可使发热病人体温下降至正常,而不影响正常人的体温。该类药物对头疼、牙疼、神经疼和关节疼等镇痛效果较好,而对外伤性及内脏平滑肌痉挛引起的绞痛无效。盐酸哌替定(杜冷丁) 全麻 中枢镇静催眠药、抗癫痫药。外周非甾体抗炎药 神经精神神经疾病治疗药神经局麻药 镇痛药 大多数的NSAID S是弱酸,其PKa值在3~4.5之间,少数较高如吡罗昔康为6.3,其弱酸性明显影响吸收、分布和贮存。一般来说,非离子型化合物的脂溶性较大,易于与生物膜脂双层结合而插入膜内,大多数NSAID S在一般的细胞内、外液(接近中性)药物分子呈解离型为多,但在胃、肾、髓质、炎症区或缺血区内,多呈非离子型,因这些部位酸度大,它们优先定位于这些酸性组织器官内,在这些部位的细胞膜上结合的药物的浓度明显高于血浆内游离的药物浓度,弱酸性的NSAZD S在胃的酸性条件下,易于穿透细胞膜,很快被吸收,吸收后很快产生药效,对慢性疾患数周内即可充分发挥药效。 2. 副作用(三方面) NSAID S对消化道有损伤,这是由于PG S对粘膜细胞具有保护作用。而NSAID S抑制了PG S的合成,可引起了溃疡出血;另一方面,它对毛细管的损伤可使血流减少和胃酸、蛋白
第五章第六节:解热镇痛药及非甾体抗炎药(答案)【炎症】
第五章:解热镇痛药及非甾体抗炎药(自考复习) 一、单项选择题(在每小题的五个备选答案中,选出一个正确答案,并将正确答案的序号 1、在阿司匹林合成中产生的可引起过敏反应的副产物是(A)。 A.乙酰水杨酸酐 B.吲哚 C.苯酚 D.水杨酸苯酯 E.乙酰水杨酸苯酯 2、吡唑酮类药物具有的药理作用是(B)。 A.镇静催眠 B.解热镇痛抗炎 C.抗菌 D.抗病毒 E.降低血压 3、扑热息痛药物中应检查( B )杂质的含量。 A.对乙酰氨基酚 B.对氨基酚 C.水杨酸 D.对氨基苯甲酸 E.醋酸 4、双氯芬酸钠属于哪类药物( B ) A.邻氨基苯甲酸类 B.吲哚乙酸类 C.芳基烷酸类 D.吡唑烷酮类 E.苯胺类 5、下列抗炎药与其结构类型不相对应的是( D )。 A.阿司匹林——水杨酸类 B.吲哚美辛——杂环芳基乙酸类 C.萘普生——杂环芳基丙酸类 D.布诺芬——吡唑酮类 6、布洛芬是( A )。 A.非甾体抗炎药 B.中枢兴奋药 C.心血管药 D.镇痛药 7、具有手性碳原子,临床上用其(+)异构体的药物是(C)。 A.安乃近 B.阿司匹林 C.萘普生 D.对乙酰氨基酚
8、在阿司匹林合成中产生的可引起过敏反应的副产物是( A )。 A.乙酰水杨酸酐 B.吲哚 C.苯酚 D.水杨酸苯酯 E.乙酰水杨酸苯酯 9、非甾体消炎药是通过抑制花生四烯酸(1),阻断(2)的合成,而显示消炎、解热、镇痛作用的( A )。 A.(1)环氧合酶(2)前列腺素 B.(1)酯氧酶(2)前列腺素 C.(1)环氧合酶(2)白三烯 D.(1)酯氧酶(2)白三烯 10.下列药物中没有消炎作用的是( B )。 A.阿司匹林 B.扑热息痛 C.萘普生 D.吲哚美辛 11、抗抑郁药盐酸氟西汀是哪种生物胺的重摄取的抑制剂?( B ) A.多巴胺 B.5-羟色胺 C.去甲肾上腺素 D.色胺 12、下列药物中,没有镇痛作用的是( C )。 A.阿司匹林 B.海洛因 C.扑热息痛 D.苯妥英钠 13、下列镇痛消炎药中,具有芳基丙酸结构的是( D )。 A.阿司匹林 B.吲哚美辛 C.保泰松 D.布洛芬 14、加FeCl3立即显稳定红色的是( D ) A.氨甲丙二酯 B.卡马西平 C.苯巴比妥 D.氯丙嗪 E.安定 15、扑热息痛药物中应检查_______杂质的含量。( B ) A.对乙酰氨基酚 B.对氨基酚 C.水杨酸 D.对氨基苯甲酸 E.醋酸 16、具有手性碳原子,临床上用其(+)异构体的药物是(C )