Western Blot图片灰度分析 -Photoshop

Western Blot图片灰度分析–Photoshop
Comparing the intensity of bands on a Western blot can be done in a number of ways using software that is commonly found on lab computers or freely available for download. The following document outlines some of those methods.
For a long time, the de-facto standard for analysis in labs that didn't want to spring for multi-thousand $$ commercial densitometry software was Adobe Photoshop or one of the competing photo editing programs. All you really need in a program is a freehand selection tool and a way to measure the mean gray value inside the selection. We'll start with Adobe Photoshop as an example (you can find many references in the literature that include phrases such as "densitometry was carried out in Photoshop" in the methods section).
To start with, you'll need to scan in your xray film on a flat-bed scanner. This can be a cheap consumer unit, or a more expensive transparency scanner if you have access to such a beast. You can scan the film as a grayscale image, and set the resolution to a medium value (300-400dpi).
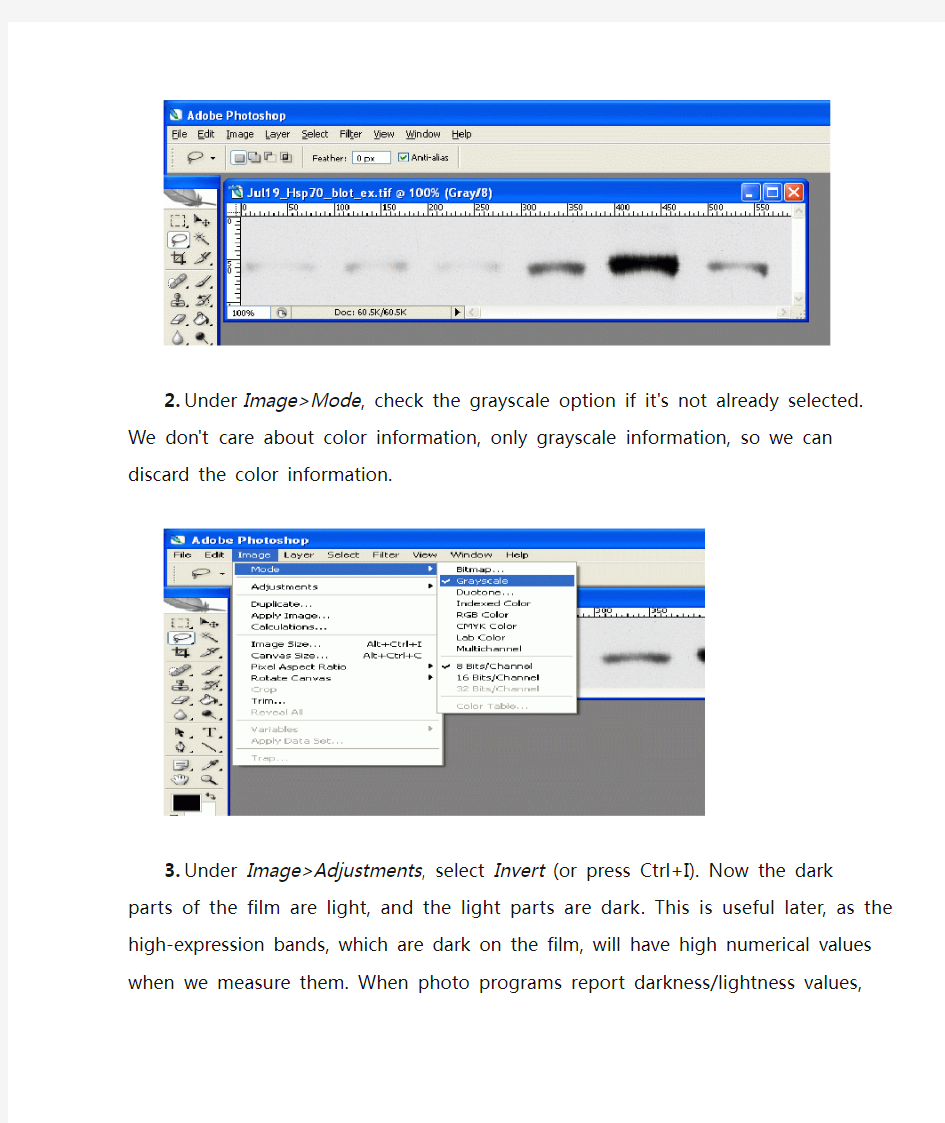
1.Open the scanned image in Photoshop.
2.Under Image>Mode, check the grayscale option if it's not already selected. We don't care
about color information, only grayscale information, so we can discard the color information.
3.Under Image>Adjustments, select Invert (or press Ctrl+I). Now the dark parts of the film
are light, and the light parts are dark. This is useful later, as the high-expression bands, which are dark on the film, will have high numerical values when we measure them.
When photo programs report darkness/lightness values, the dark points have values near zero, and the light points have values that max out at 255.
The inverted scan
4.Choose the lasso tool from the tool palette.
5.On the first band, use the lasso tool to draw a line all the way around the edges of the first
band. This is where your judgement comes in to play, determining where the edges of the
band are, and what is simply background.
6.For Photoshop CS2/3: If the histogram window is not open by default, go to
Window>Histogram to open the histogram. In newer versions of Photoshop (CS2, CS3) there is a small arrow in a circle in the upper right corner of the histogram window. Click on this and choose 'expanded view' to show the values for your selection.
For Photoshop v.5+6: Go to Image>Histogram to display the histogram for the current selection.
6.The histogram information includes a "Mean" value and a "Pixels" value. Record these
two numbers for your selection. The Mean value is the average gray value (from 0 to 255) for the area inside your selection. The Pixels value is the number of pixels contained in your selection area. Bands with high expression are typically darker, but also often larger in size, so we want to know both of these attributes for our comparison later.
8. On your picture, use the lasso tool to draw around the next band. Record the Mean and Pixel values for this selection. Repeat for the rest of your bands, including your standard.
ImageJ对WesternBlot进行灰度分析
Image J 对Western blot 条带进行灰度分析 Image J软件:Windows 版本 第一步:软件安装 1.下载地址:https://www.360docs.net/doc/1017226675.html,/ij/download.html 2.下载windows 32位带Java版本,双击软件安装。 第二步:界面介绍 1.软件界面 第三步:图片分析 1.下图为样板图片 2.导入图片:File> Open> Sample1.jpg 图片导入后,Sample1.jpg在新的窗口中打开
3.图片类型设置:Image> Type> 8 bit 4.去除图片背景:Process> Subtract Background> …
4.1Subtract Background 窗口:Rolling ball radius 设为50 pixels, 勾选light background,可选preview,点击“OK”确定 5.工具栏:选择“矩形选框”
6.最大化“Sample1.jpg”窗口,便于矩形选择,矩形选择第一个泳道 7.标记“矩形选框”为1:矩形选择后,调整矩形至合适大小,按下数字键 “1”,或者Analyze> Gels> Select Fist Lane。 8.选择“泳道2”:拖动“1”的矩形框,会出现两个矩形选框,拖动矩形框 至泳道2,调整位置,并按下数字键“2”,Image J 会自动调整大小使“矩形框2”与“矩形框1”保持同一水平。 9.继续拖动,会出项新的“矩形选框”,调增至泳道3,并按下数字键“2” (注意:是按下数字键“2”),重复9至6个泳道全部选择。 10.所有泳道选择后,按下数字键“3”,出现分析图谱。
western blot条带分析
近几天回答了蛋白版几个有关压片和显影的帖子,发现在这一块还没有比较系统的论述。特贡献了western显影常见问题及解决方案,是我的经验总结,以供大家参考。其中绝大部分问题是我所遇到过的,其他个别我没遇到过的是参照pierce公司的说明书。限于字数,语言比较简洁,如对其中内容有疑问或建议可跟帖。 Western荧光检测取决于抗原含量—一抗敏感度—二抗敏感度—底物敏感度—显影和定影效率这一系统。我的经历认为确保万无一失的做法是如果看到荧光则压10~30s,看不到则同时压两张,10~30min洗一张,如果无则再补一张,1h后洗。再无,则压过夜。共3张片,根据每次结果调整。 其中两张片子的压法如下描述: 先剪一张比膜长2~3厘米的片子(以供贴胶带),然后剪2片细小透明胶带贴到片子的左右两边(贴时胶带留一半用于粘到封口膜上),然后将片子压到膜上,并使透明胶带留出的一半粘到封口膜上。这样就固定了第一张胶片,就不会因为放、取第二张胶片而使第一张移动了。然后放第二张胶片,与第一张片子贴在一起就行,注意片子要干燥,压片前用纸擦干封口膜,以避免被底物液体污染导致与下面一张粘在一起。取时用手轻轻拂过就可以把上面片子与下面一张错开了,不要用手抠,否则下面一张也移动了。在洗下面一张片子时一定要取掉粘在片子上的胶带,否则在胶带的地方会影响显影。荧光经过下面一张胶片会稍微有些衰减,所以下面一张胶片感光稍微比上面一张强一些。 现象-- - - - - - - - - - - - - - - - - - --原因- - - - - - - - - - - - - - - - --- -- -- --- -- ---解决 反影(白色条带,黑色背景) ---- -- -1 HRP过高(背景较高) - - - - - - - - ---- - - - 至少成倍稀释一抗/二抗(可4~10倍)*注1 显影后膜上条带处变为黄色---- - -2 HRP过高(敏感性太高)- - - - - - - - - ---- -至少成倍稀释一抗/二抗(可4~10倍)*注2 信号弱或无---- - - - - - - - - - ---- 3 HRP过高引起底物迅速耗竭- - - - - -- - - -至少成倍稀释一抗/二抗(可4~10倍)*注3 --- - - - - - - - - - - - - - ---- - - - --4 抗体-抗原系统敏感性过低- --- - - - - - - 提高抗体浓度/蛋白上样量*注4 - - - - - - - - - - - - - - - ---- - - - --5 转膜不充分/过转- -- -- - - -- - - - - ---- -优化转膜条件*注5 -- - - - - - - - - - - - - - ---- - - - - -6 HRP或底物活性降低---- - - -- - - -- - ----测试活性或选用敏感底物*注6 高背景---- - - - - - - - - - - - - - - -7 HRP过高(背景较高) - -- - - - - - - - - - -- 至少成倍稀释一抗/二抗(可4~10倍)*注7 ---- - - - - - - - - - - - - - --- - - ----8 封闭时间不足- - - - - - - - - - - -- - - - -- 延长封闭时间4度过夜最好*注8 --- - - - - - - - - - - - - - --- - -- -- -9 抗体与封闭液有交叉----- - - - - - - - - - -更换封闭液(如3%BSA的TBST)*注9 --- - - - - - - - - - - - - - - -- - -- - -10 TBST洗脱不足- - - - - - - ---- - --- - - -增加洗脱时间、次数、用量*注10 ---- - - - - - - - - - - - - - -- - -- - --11 暴光时间过长- - - - - ---- - - ------ -- --降低暴光时间*注11 - - - - - - - - - - - - - - - - -- - - ----12 抗原-抗体浓度过高- - - - - - -- --- - - - 降低抗体浓度或蛋白上样量*注12
westernblot详细图解
Western免疫印迹(Western Blot)是将蛋白质转移到膜上,然后利用抗体进行检测的方法。对已知表达蛋白,可用相应抗体作为一抗进行检测,对新基因的表达产物,可通过融合部分的抗体检测。 与Southern或Northern杂交方法类似,但Western Blot采用的是聚丙烯酰胺凝胶电泳,被检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。 经过PAGE分离的蛋白质样品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以
检测电泳分离的特异性目的基因表 达的蛋白成分。该技术也广泛应用 于检测蛋白水平的表达。 实验材料蛋白质样品 试剂、试剂盒丙烯酰胺SDS Tris-HCl β-巯基乙醇 ddH2O 甘氨酸Tris 甲醇PBS NaCl KCl Na2HPO4 KH2PO4 ddH2O 考马斯亮兰乙酸脱脂奶粉硫酸镍胺H2O2 DAB试剂盒 仪器、耗材 电泳仪电泳槽离心机离心管硝酸纤维素 膜匀浆器剪刀移液枪刮棒 实验步骤一、试剂准备 1. SDS-PAGE试剂:见聚丙烯酰胺凝胶电泳实验。 2. 匀浆缓冲液:1.0 M Tris-HCl(pH 6.8) 1.0 ml;10%SDS 6.0 ml;β-巯基乙醇0.2 ml;ddH2O 2.8 ml。 3. 转膜缓冲液:甘氨酸2.9 g;Tris 5.8 g;SDS 0.37 g;甲醇200 ml;加ddH2O定容至1000 ml。
Western Blot常见问题及处理总结
免疫细胞研究western blot Western Blot常见问题及处理总结 阿木 1、western blot 的优点 答:灵敏,可达ng级,用Ecl显色法理论上可达pg 级。方便,特异性高。 2、为什么我的细胞提取液中没有目标蛋 白? 答:原因有很多: a) 你的细胞中不表达这种蛋白质,换一种细胞;b) 你的细胞中的蛋白质被降解掉了,你必需加入PMSF,抑制蛋白酶活性;c) 你的抗体不能识别目标蛋白,多看看说明,看是否有问题。 3、我的细胞提取液有的有沉淀,有的很 清亮,为什么呢?
答:a) 有沉淀可能因为你的蛋白没有变性完全,可以适当提高SDS 浓度,同时将样品煮沸时间延长,b) 也不排除你的抗原浓度过高,这时再加入适量上样缓冲 液即可。 4、我做的蛋白质分子量很小(10KD), 请问怎么做WB? 答:可以选择0.2μml的膜,同时缩短转移时间。也可以将两张膜叠在一起,再转移。 其他按步骤即可。 5、我的目的带很弱,怎么加强? 答:可以加大抗原上样量。这是最主要的。 同时也可以将一抗稀释比例降低。 6、胶片背景很脏,有什么解决方法?答:减少抗原上样量,降低一抗浓度,改
变一抗孵育时间,提高牛奶浓度。 7、目标带是空白,周围有背景,是为什 么? 答:你的一抗浓度较高,二抗上HRP 催化活力太强,同时你的显色底物处于一个临界点,反应时间不长,将周围底物催化完,形成了空白即“反亮现象”。将一抗和二抗浓度降低,或更换新底物。 8、我的胶片是一片空白,是怎么回事? 答:如果能够排除下面的几个问题那么问题多半出现在一抗和抗原制备上。 a) 二抗的HRP 活性太强,将底物消耗光;b) ECM底物中H2O2,不稳定,失活;c) ECL底物没覆盖到相应位置;d) 二 抗失活。
WesternBlot结果条带分析
W e s t e r n B l o t结果条带 分析 Revised final draft November 26, 2020
W e s t e r n B l o t结果条带全面分析 1.空白 原因:比较多,如果单纯一张没有任何显色,最可能是一抗加成其他抗体,或者二抗种属加错了,比如兔的加成鼠的。 解决办法:仔细检查抗体是否加错,确认转膜没有问题。上面的图片展示的是一点信号都没有,如果是这样大部分情况是抗体加错了。如果中间出现了细微的条带,可能原因是蛋白上样量太少,一抗浓度过低,显色液失效。另外如果转膜出现了问题,比如膜放反了,自然是一个白片。 2.高背景 原因:封闭不够好,一抗浓度高,洗膜时间和次数不够。 解决办法:降低一抗浓度,增加洗膜时间和次数。 3.非特异性条带 原因:一抗非特异性与蛋白结合 解决办法:更换一抗 4.条带中出现边缘规则的白圈 原因:电转中膜和胶之间存在气泡。 解决办法:转膜前去掉膜和胶之间的气泡 5.出现黑点和黑斑 原因:膜上其他部位与一抗或者二抗非特异性结合 解决办法:封闭结束之后要洗。 6.条带拖尾 原因:蛋白量太大,一抗浓度和时间太长 解决办法:根据情况调整蛋白量,同时一抗浓度和时间也可以缩短。 7.出现非均一性背景 原因:膜可能曾经干过 解决办法:在每一步的操作过程中,都需要注意不要让膜干。 8.某个条带变形 原因:SDSPAGE胶中存在气泡或者某不溶性颗粒 解决办法:配胶过程中要小心,使用无杂质的液体。另外配胶用的水,SDS,Tris缓冲液要注意不要有杂质。 9.条带呈哑铃状 原因:配置胶有问题,胶凝固后不均一 解决办法:把胶配好,不合格的胶坚决不用 10.最边缘条带弯曲 原因:电泳电流不均一 解决办法:换用新的电泳槽;不使用两边的两孔 其他问题 (1)蛋白分子量偏高或者偏低。可能是胶的浓度与目的蛋白的浓度不对应。 (2)蛋白质降解。蛋白质降解后很可能会在比原来位置低的地方出现主带,然后会出现一些其他带,最主要特点是所有的条带比正常的都低,并且条带模糊不清晰。 (3)所有条带连成一片没有间隔。原因最可能是上样量过多,其次是样品弥散(比如电泳长时间停止样品弥散)。 (4)整个条带呈“︶”状:凝胶冷却不均一,电泳槽老化。 (5)整个条带呈“︵”:凝胶左右两头没有凝固好 (6)溴酚蓝拖尾:样品溶解不好。 (7)纵向的纹理:上样样品中存在不溶性颗粒 (8)溴酚蓝很粗:浓缩胶浓缩效果不好,可能是浓缩胶太短,或者是浓缩胶配错。
western blot实验经验总结
western blot实验经验总结 1.抗体的选择 对于国内的大多数实验室来讲,做western blot实验选择抗体是个头疼的问题。原因很简单,买进口抗体捉襟见肘,买国产抗体得需要大无畏的勇气,对于我所在的兰州地区的实验者而言,感触尤深。在这五年的western blot实验历程里,我先后用过进口抗体,进口抗体国内分装包装,国产抗体,质量良莠不齐。 进口抗体一般不会出现闪失:abcam品种全,质量过硬,但价高(3400元/100微升),而且说是100微升,但至多能吸出来90微升;Sigma的价最贵;比较有性价比的是CST的抗体,现在好像是2200元/100微升,我前后用过二十几种, 1:1000的稀释比下,还没有失过手,100微升通常能完成所有的免疫组化和western blot 实验,还有一个优点是通常量比100微升多出来10微升,唯一的不足是CST的品种实在不多。Santa的多克隆抗体质量可以,但是选用Santa的单抗还是有风险,估计这也是业界共识了吧。 进口抗体国内分装包装我也用过不少,呵呵,毕竟是穷人(420元/100微升),大概好抗体的比例约为50%,如果能做出来,也存在一个问题,就是抗体大多只能用一次。我曾经把Santa的原装抗体和分装抗体做过比对(抗体品种,货号等完全一致),在都能做出来的前提下,原装抗体能重复使用的次数要多出许多。具体原因我已经揣测了好几年,不敢说出来,我一直在想是不是冬天西瓜切成牙和整个卖,也会有所不同。揣测归揣测,假如只为了发论文毕业走人,可以考虑选用进口分装的多克隆抗体。 国产抗体比较知名的就几家,但质量确实不敢恭维。western blot能做出来的确实不多,而且杂带多,背景不干净。我们周边的实验室大多买国产抗体做免疫组化,怎么说呢,应付硕士论文够了。 我也帮别人自制过抗体,再用抗原亲和纯化。效价非常不错,夸张的时候1:10000都能做出条带,唯一的麻烦是兔子太骚(骚臭),不知道这是不是“兔女郎”名号的来由。如果读书非常悠闲,老板又特别想拥有手工作坊的情况下,可以自己伺候折腾兔子来玩玩,刚开始的时候还是蛮有成就感的。只是单凭抗原表达和制备多抗,是不是可以写成论文毕业,估计因校而异了。 2.Western Blot设备 目前最好的垂直槽和转移槽还是Biorad(伯乐),尤其是MINI3好用,MINI4可以一次跑四块胶,但通常用不上,由此造成垂直缓冲液的浪费,而且MINI4用绿色塑料夹住玻璃板来组成内槽,容易漏液,塑料夹应该是有疲劳寿命的,估计日子一长,弹性就会改变,所以如果你们实验室刚买了MINI4,赶紧用,遭殃的肯定是某一级的师弟师妹们。 Biorad的一套系统得两万多,西部能玩得起伯乐的还真不多。好在咱中国人聪明,上海天能的外观和构造和伯乐的MINI3几乎一模一样,而且可以通用,不带电泳仪是5千多一套,挺好用的。唯一的不足是塑料寿命不如伯乐,胶架容易断裂。不过仔细算算,三套天能也就是一套伯乐的价格,值了。北京六一的垂直槽
蛋白质印迹法westernblot
蛋白质印迹法 蛋白质印迹法(免疫印迹试验)即Western Blot。它是分子生物学、生物化学和免疫遗传学中常用的一种实验方法。 其基本原理是通过特异性抗体对凝胶电泳处理过的细胞或生物组织样品进行着色。通过分析着色的位置和着色深度获得特定蛋白质在所分析的细胞或组织中表达情况的信息。 蛋白免疫印迹(Western Blot )是将电泳分离后的细胞或组织总蛋白质从凝胶转移到固相支持物NC膜或PVDF膜上,然后用特异性抗体检测某特定抗原的一种蛋白质检测技术,现已广泛应用于基因在蛋白水平的表达研究、抗体活性检测和疾病早期诊断等多个方面。 中文名蛋白质印迹法外文名Western Blot 蛋白免疫印迹Western Blot 类似方法1 Southern Blot 杂交方法 类似方法2 Northern Blot 杂交方法 使用材料聚丙烯酰氨凝胶电泳⑴
原理 与Southern Blot 或Northern Blot 杂交方法类似,但Western Blot法采用的是聚丙烯酰胺凝胶电泳,被检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。经过PAG(聚丙烯酰胺凝胶电泳)分离的蛋白质样品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。该技术也广泛应用于检测蛋白水平的表达。⑴ 分类 Western Blot 显色的方法主要有以下几种: i. 放射自显影 ii. 底物化学发光ECL iii. 底物荧光ECF iv. 底物DAB呈色
westernblot详细图解详细版.docx
Western免疫印迹(Western Blot) 是将蛋白质转移到膜上,然后利用 抗体进行检测的方法。对已知表达 蛋白,可用相应抗体作为一抗进行 检测,对新基因的表达产物,可通 过融合部分的抗体检测。 与Southern或Northern杂交方法 类似,但Western Blot采用的是聚 丙烯酰胺凝胶电泳,被检测物是蛋 白质,“探针”是抗体,“显色”用标记 的二抗。 经过PAGE分离的蛋白质样品,转 移到固相载体(例如硝酸纤维素薄 膜)上,固相载体以非共价键形式 吸附蛋白质,且能保持电泳分离的 多肽类型及其生物学活性不变。以 固相载体上的蛋白质或多肽作为抗 原,与对应的抗体起免疫反应,再 与酶或同位素标记的第二抗体起反 应,经过底物显色或放射自显影以 检测电泳分离的特异性目的基因表 达的蛋白成分。该技术也广泛应用 于检测蛋白水平的表达。 实验材料蛋白质样品 试剂、试剂盒丙烯酰胺SDS Tris-HCl β-巯基乙醇 ddH2O 甘氨酸Tris 甲醇PBS NaCl KCl Na2HPO4 KH2PO4 ddH2O 考马斯亮兰乙酸脱脂奶粉硫酸镍胺H2O2 DAB试剂盒 仪器、耗材电泳仪电泳槽离心机离心管硝酸纤维素膜匀浆器剪刀移液枪刮棒 实验步骤一、试剂准备 1. SDS-PAGE试剂:见聚丙烯酰胺凝胶电泳实验。 2. 匀浆缓冲液:1.0 M Tris-HCl(pH 6.8) 1.0 ml;10%SDS 6.0 ml;β-巯基乙醇0.2 ml;ddH2O 2.8 ml。 3. 转膜缓冲液:甘氨酸2.9 g;Tris 5.8 g;
SDS 0.37 g;甲醇200 ml;加ddH2O定容至1000 ml。 4. 0.01 M PBS(pH7.4):NaCl 8.0 g;KCl 0.2 g;Na2HPO4 1.44 g;KH2PO4 0.24 g;加ddH2O至1000 ml。 5. 膜染色液:考马斯亮兰0.2 g;甲醇80 ml;乙酸2 ml;ddH2O118 ml。包被液(5%脱脂奶粉,现配):脱脂奶粉1.0 g 溶于20 ml的0.01 M PBS中。 6. 显色液:DAB 6.0 mg;0.01 M PBS 10.0 ml;硫酸镍胺0.1 ml;H2021.0 μl。 二、蛋白样品制备 1. 单层贴壁细胞总蛋白的提取 (1)倒掉培养液,并将瓶倒扣在吸水纸上使吸水纸吸干培养液(或将瓶直立放置一会儿使残余培养液流到瓶底然后再用移液器将其吸走)。 (2)每瓶细胞加3 ml 4℃预冷的PBS (0.01M pH7.2~7.3)。平放轻轻摇动1 min 洗涤细胞,然后弃去洗液。重复以上操作两次,共洗细胞三次以洗去培养液。将PBS弃净后把培养瓶置于冰上。 (3)按1ml裂解液加10 μl PMSF(100 mM),摇匀置于冰上。(PMSF要摇匀至无结晶时才可与裂解液混合。) (4)每瓶细胞加400 μl含PMSF的裂解液,于冰上裂解30 min,为使细胞充分裂解培养瓶要经常来回摇动。 (5)裂解完后,用干净的刮棒将细胞刮于培养瓶的一侧(动作要快),然后用枪将细胞碎片和裂解液移至1.5 ml离心管中。(整个操作尽量在冰上进行。) (6)于4℃下12000 rpm离心5 min。(提
western-blot条带分析及解决方法
*注1其具体问题有二:一是背景较高,二是没有条带?形成机制为条带处HRP很快将底物耗竭,则不发光不使胶片感光而为白色,周围因背景H R P较低而持续发光一段时间使胶片感光为黑色。这种常见于高浓度抗体并且封闭/洗脱不好的情况下。如果没有背景则就是原因3的现象。在暗室可以看到
条带周围荧光而条带处为暗。如是则要么通过降低抗体解决,要么动作迅速早期未耗竭底物时压片,否则一旦耗竭通过减少压片时间不能解决问题。也可以过一段时间鼿RP稍减后重加底物迅速压片。还有另外一种反影现象就是压出条带粗大模糊,但条带中心是空的白色,原因和上面的一样,主要是条带中心耗竭底物引起的,也可以在暗室看到中空的荧光带,但与上面的区别主要在于特异性要好一些,若继续发展则就是原因3的现象。其分析和解决同上。 *注2其原因主要是HRP太高超速催化底物生成的产物使膜发黄,压出的片子可以是正常的或和原因1或原因3的现象同时存在,只是一种现象,在压过的膜可以看出来(只在加底物后一段时间出现,几小时后可能还会消退,有时甚至刚加底物1~2min就可以看出来,所以要把握时机)。如果没有 达到原因1和原因3的程度,那么这种情况一定能够看到很强的荧光,是一种良好的状态,是我们所期盼的。如果压出正常片子而不出现原因1和原因3的现象,可以不予处理。但因为荧光太强极易导致条带增粗变大,所以也可以不稀释抗体而通过调整压片时间来解决,一般压10s~30s,甚至可以3~5s,即时根据结果在暗室调整,荧光持续时间长的话都可连续压十数张片子而效果良好。但也有这种情况,就是由于HRP过强无论怎么减少压片时间条带都依然粗大(如果时间过短如<3s则可因感光不足导致条带太淡,这样就不可取了,但依然是粗大的、非条带的本来面目),那么如果想获得漂亮条带则必须通过降低抗体浓度(减少上样量很不稳定且能力有限,故很少使用)来解决。原因1和3若如是则解决也是一样的。 *注3这是与1类似但抗体特异性较好时的一种表现,经常与下面的原因4、5、6混淆,特别需要注意。主要出现在HRP极高的情况下,来不及压片就已耗竭底物,往往伴有原因2的现象。可通过加入底物后迅速进入暗室观察荧光确定,也可以根据原因2的现象确定,以区分于原因4、5、6。 除非动作迅速早期未耗竭底物时压片,否则一旦耗竭通过减少压片时间不能解决问题。也可以过一段时间待HRP稍减后TBST简单洗膜重新加底物压片。这种情况下一/二抗稀释可高达10倍而依然荧光明显。 *注4提高抗体上样量以及下述的使用敏感底物/延长压片时间比较简单,在此不赘述,只提醒一下随抗体浓度增高背景和非特异性可能会增加。关于上样量的极限在此发表我的看法:对于裂解的混合蛋白以上样孔底面积(平方毫米)乘30ug作为极限上样量,而落实到每一个具体条带(同一分子量的 所有蛋白或仅做单一蛋白电泳),则以0.3ug/平方毫米底面积作为极限上样量。(见《抗体技术实验指南》的免疫印迹一章),超过此量可能会导致条带变形。条带信号弱可以通过加大上样量/提高抗体浓度/使用敏感底物/延长压片时间解决。但实际上样量的极限并不以我说的极限上样量为准,因为这个极限上样量是确保所有分子量的条带都跑的很漂亮的极限,而上样超量的表现是随量的加大变形的条带由低分子量逐渐往高分子量延伸。 所以western极限上样量的真正操作标准是以目的条带不变形作为极限。比如对于150kd,完全可以超出我所说的极限上样量的2倍而不变形(此时中低分子量条带早已融合或挤为棒槌形了)
Westernblot实验步骤及注意事项
Westernblot实验步骤及注意事项 Westernblot 实验步骤 1. 组织块称重 2. 利用液氮、研钵粉碎组织块 3. 加入RIPA缓冲液(每克组织3 ml RIPA),PMSF(每克组织30μl,10 mg/ml PMSF),利用Polytron进一步匀浆(15,000转/分*1分钟)维持4℃ 4. 加入PMSF(每克组织30μl,10 mg/ml PMSF),冰上孵育30分钟 5. 移入离心管4℃约20,000 g(约15,000转)15分钟 6. 上清液为细胞裂解液可分装-20℃保存 7. 进行Bradford比色法测定蛋白质浓度 8. 取相同质量的细胞裂解液(体积*蛋白质浓度),并加等体积的2×电泳加样缓冲液 9. 沸水浴中3分钟 10. 上样 11. 电泳(浓缩胶20mA,分离胶35mA) 12. 电转膜仪转膜(100mA 40分钟) 13. 膜用丽春红染色,胶用考马斯亮蓝染色 14. Westernblot 试剂盒显色 15. 分析比较记录 western blot的实验步骤及注意事项的资料 1. 把聚丙烯酰胺凝胶中的蛋白质电泳转移到硝酸纤维膜上。 1)转移缓冲液洗涤凝胶和硝酸纤维素膜,将硝酸纤维素膜铺在凝胶上,用5ml移液管在凝胶上来回滚动去除所有的气泡。 2)在凝胶/滤膜外再包一张3mm滤纸(预先用转移缓冲液浸湿),将凝胶夹在中间,保持湿润和没有气泡。薄膜滤纸按照厂家建议方法放入电泳装置中,凝胶面向阴极。/凝胶/)将此滤纸3.
4)将上述装置放入缓冲液槽中,并灌满转移缓冲液以淹没凝胶。 5)按照厂家所示接通电源开始电泳转移。 6)转移结束后,取出薄膜和凝胶,弃去凝胶。 2. 将薄膜漂在氨基黑中快速染色,直至分子量标准显现时取出,记录下标准位置。 3. 用100ml水洗涤纤维素膜,必要时可用脱色缓冲液。 4. 膜置印迹缓冲液中于37℃保温1小时。 5. 室温下,用PBS-Tween缓冲液洗涤薄膜。 6. 用封口机将薄膜封入塑料袋中,尽可能不留空气。 7.袋的一角剪一缓冲液的小口,用透析袋夹紧。 8.混合:NGS(100微升),印迹缓冲液中的抗体(10毫升),加在装薄膜的袋中,于室温下摇动2小时(或4℃过夜) 9.用总体积300ml PBS-Tween缓冲液,分4次在一浅盘中洗涤薄膜,每次75ml。 10.将连接生物素的羊抗兔IgG(40微升溶于10毫升印迹缓冲液/100微升NGS)加在袋内,于室温下摇动1小时。 11.按步骤9洗涤。 12.加入抗生素蛋白-HRP(40微升溶于10毫升印迹缓冲液/100微升NGS),于室温下摇动。 注意事项: western blot中转移在膜上的蛋白处于变性状态,空间结构改变,因此那些识别空间表位的抗体不能用于western blot检测。这种情况可以将表达目的蛋白的细胞或细胞裂解液中的所有蛋白先生物素化,再用酶标记亲和素进行western blot。实验中取胶和膜需带手套。 Western实验步骤 Western,也称Western blot、Western blotting、Western印迹,是用抗体检测蛋白的重要方法之一。Western 可以参 考如下步骤进行操作。 1. 收集蛋白样品(Protein sample preparation) O 可以使用适当的裂解液,例如碧云天生产的Western及IP细胞裂解液,裂解贴壁细胞、悬浮细胞或组织样品。对于某些特定的亚 细胞组份蛋白,例如细胞核蛋白、细胞浆蛋白、线粒体蛋白等,可以参考相关文献提取这些亚细胞组份蛋白,也可以使用试剂盒 进行抽提,例如碧云天生产的细胞核蛋白与细胞浆蛋白抽提试剂盒。 O 收集完蛋白样品后,为确保每个蛋白样品的上样量一致,需要测定每个蛋白样品的蛋白浓度。根据所使用的裂解液的不同,需要 采用适当的蛋白浓度测定方法。因为不同的蛋白浓度测定方法对于一些去垢剂和还原剂等的兼容性差别很大。如果使用碧云天生
western-blot条带分析报告及解决方法
持续发光一段时间使胶片感光为黑色。这种常见于高浓度抗体并且封闭/洗脱不好的情况下。如果没有背景则就是原因3的现象。在暗室可以看到文案大全
条带周围荧光而条带处为暗。如是则要么通过降低抗体解决,要么动作迅速早期未耗竭底物时压片,否则一旦耗竭通过减少压片时间不能解决问题。也可以过一段时间鼿RP稍减后重加底物迅速压片。还有另外一种反影现象就是压出条带粗大模糊,但条带中心是空的白色,原因和上面的一样,主要是条带中心耗竭底物引起的,也可以在暗室看到中空的荧光带,但与上面的区别主要在于特异性要好一些,若继续发展则就是原因3的现象。其分析和解决同上。 *注2 其原因主要是HRP太高超速催化底物生成的产物使膜发黄,压出的片子可以是正常的或和原因1或原因3的现象同时存在,只是一种现象,在压过的膜可以看出来(只在加底物后一段时间出现,几小时后可能还会消退,有时甚至刚加底物1~2min就可以看出来,所以要把握时机)。如果没有达到原因1和原因3的程度,那么这种情况一定能够看到很强的荧光,是一种良好的状态,是我们所期盼的。如果压出正常片子而不出现原因1和原因3的现象,可以不予处理。但因为荧光太强极易导致条带增粗变大,所以也可以不稀释抗体而通过调整压片时间来解决,一般压10s~30s,甚至可以3~5s,即时根据结果在暗室调整,荧光持续时间长的话都可连续压十数张片子而效果良好。但也有这种情况,就是由于HRP过强无论怎么减少压片时间条带都依然粗大(如果时间过短如<3s则可因感光不足导致条带太淡,这样就不可取了,但依然是粗大的、非条带的本来面目),那么如果想获得漂亮条带则必须通过降低抗体浓度(减少上样量很不稳定且能力有限,故很少使用)来解决。原因1和3若如是则解决也是一样的。 *注3 这是与1类似但抗体特异性较好时的一种表现,经常与下面的原因4、5、6混淆,特别需要注意。主要出现在HRP极高的情况下,来不及压片就已耗竭底物,往往伴有原因2的现象。可通过加入底物后迅速进入暗室观察荧光确定,也可以根据原因2的现象确定,以区分于原因4、5、6。除非动作迅速早期未耗竭底物时压片,否则一旦耗竭通过减少压片时间不能解决问题。也可以过一段时间待HRP稍减后TBST简单洗膜重新加底物压片。这种情况下一/二抗稀释可高达10倍而依然荧光明显。 *注4 提高抗体上样量以及下述的使用敏感底物/延长压片时间比较简单,在此不赘述,只提醒一下随抗体浓度增高背景和非特异性可能会增加。关于上样量的极限在此发表我的看法:对于裂解的混合蛋白以上样孔底面积(平方毫米)乘30ug作为极限上样量,而落实到每一个具体条带(同一分子量的所有蛋白或仅做单一蛋白电泳),则以0.3ug/平方毫米底面积作为极限上样量。(见《抗体技术实验指南》的免疫印迹一章),超过此量可能会导致条带变形。条带信号弱可以通过加大上样量/提高抗体浓度/使用敏感底物/延长压片时间解决。但实际上样量的极限并不以我说的极限上样量为准,因为这个极限上样量是确保所有分子量的条带都跑的很漂亮的极限,而上样超量的表现是随量的加大变形的条带由低分子量逐渐往高分子量延伸。所以western极限上样量的真正操作标准是以目的条带不变形作为极限。比如对于150kd,完全可以超出我所说的极限上样量的2倍而不变形(此时中低分子量条带早已融合或挤为棒槌形了) 文案大全
最详细的WesternBlot过程步骤详解
最详细的W e s t e r n B l o t 过程步骤详解
最详细的W e s t e r n B l o t 过程步骤详解 Pleasure Group Office【T985AB-B866SYT-B182C-BS682T-STT18】
Western Blot详解(原理、分类、试剂、步骤及问题解答) Western免疫印迹(Western Blot)是将蛋白质转移到膜上,然后利用抗体进行检测。对已知表达蛋白,可用相应抗体作为一抗进行检测,对新基因的表达产物,可通过融合部分的抗体检测。 本文主要通过以下几个方面来详细地介绍一下Western Blot技术: 一、原理 二、分类 i.放射自显影 ii.底物化学发光ECL ECF iv.底物DAB呈色 三、主要试剂 四、主要步骤 五、实验常见的问题指南 1.参考书推荐 2.针对样品的常见问题 3.抗体 4.滤纸、胶和膜的问题 的相关疑问 6.染色的选择 7.参照的疑问
8.缓冲液配方的常见问题 9.条件的摸索 10.方法的介绍 11.结果分析 一、原理 与Southern或Northern杂交方法类似,但Western Blot采用的是聚丙烯酰胺凝胶电泳,被检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。经过PAGE分离的蛋白质样品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。该技术也广泛应用于检测蛋白水平的表达。 二、分类 现常用的有底物化学发光ECL和底物DAB呈色,体同水平和实验条件的是用第一种方法,目前发表文章通常是用底物化学发光ECL。只要买现成的试剂盒就行,操作也比较简单,原理如下(二抗用HRP标记):反应底物为过氧化物+鲁米诺,如遇到HRP,即发光,可使胶片曝光,就可洗出条带。
WB实验问题
Western Blot问题指南 时间:2011-05-13 08:55 来源:网络作者:admin点击:3679次 根据问题的类型主要分成以下几类(以下资料权作参考,请勿盲目模仿!); 1.参考书 推荐A.对初学者看什么资料比较好?解答:《抗体技术实验指南》和Antibodies (a laboratory manual , 根据问题的类型主要分成以下几类(以下资料权作参考,请勿盲目模仿!); 1. 参考书推荐 A. 对初学者看什么资料比较好? 解答:《抗体技术实验指南》和Antibodies (a laboratory manual,wrote by Ed Harlow ,david lane )两本书不错。 2. 针对样品的常见问题 B. 做线粒体膜UCP蛋白的Western Blot (以下简写成Western Blot),提取线粒体后冻存(未加蛋白酶抑制剂),用的博士德的一抗,开始还有点痕迹,现在越来越差,上样量已加到120 口g,换了个santa cloz 的一抗仍不行。是什么原因?蛋白酶抑制剂单力口PMSF亍吗? 解答:怀疑是样品问题,可能是:1,样品不能反复冻融;2,样品未加蛋白酶抑制剂。同时,建议检查Western Blot过程,提高一抗浓度。对于加蛋白酶抑制剂来说,一般加PMSF就可以了,最好能多加几中种蛋白酶抑制剂。 C. 同一蛋白样品能同时进行两种因子的Western Blot检测吗? 解答:当然可以,有的甚至可以同时测几十种样品。 D. 如果目标蛋白是膜蛋白或是胞浆蛋白,操作需要注意什么? 解答:如果是膜蛋白和胞浆蛋白,所用的去垢剂就要温和得多,这时最好加上NaF去抑 制磷酸化酶的活性。 E我的样品的蛋白含量很低,每微升不到1微克,但是在转膜时经常会发现只有一部分 蛋白转到了膜上,就是在转膜后染胶发现有的孔所有的蛋白条带都在,只是颜色变淡了, 有什么办法可以解决? 解答:你可以加大上样量,没有问题,还有转移时你可以用减少电流延长时间,多加5—10%甲醇。 F. 想分离的蛋白是分子量260kd的,SDS-PAGE电泳的分离胶浓度多大合适?积层胶的 浓度又该用多少?这么大分子量的蛋白容易作Wester n Blot吗? 解答:260kd的蛋白不好做,分离胶用6%, Stacking Gel 3.5 %。 G. 如果上样量超载,要用什么方法来增加上样量?如果需要加大上样量使原来弱的条带能看清楚。 解答:可以浓缩样品,也可以根据你的目标分子量透析掉一部分小分子蛋白。一般地,超载30%是不会有问题的。如果已经超了不少了,而且小分子量的也要,可以考虑加大胶的厚度,可以试试1.5m m的comb。
Western Blot结果条带分析
Western Blot结果条带全面分析 1.空白 原因:比较多,如果单纯一张没有任何显色,最可能是一抗加成其他抗体,或者二抗种属加错了,比如兔的加成鼠的。 解决办法:仔细检查抗体是否加错,确认转膜没有问题。上面的图片展示的是一点信号都没有,如果是这样大部分情况是抗体加错了。如果中间出现了细微的条带,可能原因是蛋白上样量太少,一抗浓度过低,显色液失效。另外如果转膜出现了问题,比如膜放反了,自然是一个白片。 2.高背景 原因:封闭不够好,一抗浓度高,洗膜时间和次数不够。 解决办法:降低一抗浓度,增加洗膜时间和次数。 3.非特异性条带 原因:一抗非特异性与蛋白结合 解决办法:更换一抗 4.条带中出现边缘规则的白圈 原因:电转中膜和胶之间存在气泡。 解决办法:转膜前去掉膜和胶之间的气泡 5.出现黑点和黑斑 原因:膜上其他部位与一抗或者二抗非特异性结合 解决办法:封闭结束之后要洗。 6.条带拖尾 原因:蛋白量太大,一抗浓度和时间太长 解决办法:根据情况调整蛋白量,同时一抗浓度和时间也可以缩短。 7. 出现非均一性背景 原因:膜可能曾经干过 解决办法:在每一步的操作过程中,都需要注意不要让膜干。 8.某个条带变形 原因:SDSPAGE胶中存在气泡或者某不溶性颗粒 解决办法:配胶过程中要小心,使用无杂质的液体。另外配胶用的水,SDS,Tris 缓冲液要注意不要有杂质。 9. 条带呈哑铃状 原因:配置胶有问题,胶凝固后不均一 解决办法:把胶配好,不合格的胶坚决不用 10.最边缘条带弯曲 原因:电泳电流不均一
解决办法:换用新的电泳槽;不使用两边的两孔 其他问题 (1)蛋白分子量偏高或者偏低。可能是胶的浓度与目的蛋白的浓度不对应。(2)蛋白质降解。蛋白质降解后很可能会在比原来位置低的地方出现主带,然后会出现一些其他带,最主要特点是所有的条带比正常的都低,并且条带模糊不清晰。 (3)所有条带连成一片没有间隔。原因最可能是上样量过多,其次是样品弥散(比如电泳长时间停止样品弥散)。 (4)整个条带呈“ ︶” 状:凝胶冷却不均一,电泳槽老化。 (5)整个条带呈“ ︵”:凝胶左右两头没有凝固好 (6)溴酚蓝拖尾:样品溶解不好。 (7)纵向的纹理:上样样品中存在不溶性颗粒 (8)溴酚蓝很粗:浓缩胶浓缩效果不好,可能是浓缩胶太短,或者是浓缩胶配错。 (9)在分离胶中跑不动:Tris-HCl PH值不对,或者忘记加SDS。
Western Blot常见问题及处理总结
免疫细胞研究westernblot WesternBlot常见问题及处理总结 阿木 1、westernblot的优点 答:灵敏,可达ng级,用Ecl显色法理论上可达pg级。方便,特异性高。 2、为什么我的细胞提取液中没有目标蛋白? 答:原因有很多:a)你的细胞中不表达这种蛋白质,换一种细胞;b)你的细胞中的蛋白质被降解掉了,你必需加入PMSF,抑制蛋白酶活性;c)你的抗体不能识别目标蛋白,多看看说明,看是否有问题。 3、我的细胞提取液有的有沉淀,有的很清亮,为什么呢? 答:a)有沉淀可能因为你的蛋白没有变性完全,可以适当提高SDS浓度,同时将样品煮沸时间延长,b)也不排除你的抗原浓度过高,这时再加入适量上样缓冲液即可。 4、我做的蛋白质分子量很小(10KD),请问怎么做WB? 答:可以选择0.2μml的膜,同时缩短转移时间。也可以将两张膜叠在一起,再转移。其他按步骤即可。
5、我的目的带很弱,怎么加强? 答:可以加大抗原上样量。这是最主要的。同时也可以将一抗稀释比例降低。 6、胶片背景很脏,有什么解决方法? 答:减少抗原上样量,降低一抗浓度,改变一抗孵育时间,提高牛奶浓度。 7、目标带是空白,周围有背景,是为什么?答:你的一抗浓度较高,二抗上HRP催化活力太强,同时你的显色底物处于一个临界点,反应时间不长,将周围底物催化完,形成了空白即“反亮现象”。将一抗和二抗浓度降低,或更换新底物。 8、我的胶片是一片空白,是怎么回事? 答:如果能够排除下面的几个问题那么问题多半出现在一抗和抗原制备上。 a)二抗的HRP活性太强,将底物消耗光;b)ECM 底物中H2O2,不稳定,失活;c)ECL底物没覆盖到相应位置;d)二抗失活。 9、我在显影液中显影1分钟和5分钟后,底片漆黑一片,是什么原因呢? 答:a)可能是红灯造成的,胶片本来就被曝光了,可以在完全黑暗的情况下操作.看是否有改善.; b)显影时间过长。 10、DAB好还是ECM好?
western-blot聚丙烯酰胺凝胶电泳的基本原理及常见问题分析复习课程
聚丙烯酰胺凝胶电泳的基本原理及常见问题分析 聚丙烯酰胺凝胶电泳简称为PAGE(Polyacrylamide gel electrophoresis),是以聚丙烯酰胺凝胶作为支持介质的一种常用电泳技术。聚丙烯酰胺凝胶由单体丙烯酰胺和甲叉双丙烯酰胺聚合而成,聚合过程由自由基催化完成。催化聚合的常用方法有两种:化学聚合法和光聚合法。化学聚合以过硫酸铵(AP)为催化剂,以四甲基乙二胺(TEMED)为加速剂。在聚合过程中,TEMED催化过硫酸铵产生自由基,后者引发丙烯酰胺单体聚合,同时甲叉双丙烯酰胺与丙烯酰胺链间产生甲叉键交联,从而形成三维网状结构。 PAGE根据其有无浓缩效应,分为连续系统和不连续系统两大类,连续系统电泳体系中缓冲液pH值及凝胶浓度相同,带电颗粒在电场作用下,主要靠电荷和分子筛效应。不连续系统中由于缓冲液离子成分,pH,凝胶浓度及电位梯度的不连续性,带电颗粒在电场中泳动不仅有电荷效应,分子筛效应,还具有浓缩效应,因而其分离条带清晰度及分辨率均较前者佳。不连续体系由电极缓冲液、浓缩胶及分离胶所组成。浓缩胶是由AP催化聚合而成的大孔胶,凝胶缓冲液为pH6.7的Tris-HC1。分离胶是由AP催化聚合而成的小孔胶,凝胶缓冲液为pH8.9 Tris-HC1。电极缓冲液是pH8.3 Tris-甘氨酸缓冲液。2种孔径的凝胶、2种缓冲体系、3种pH值使不连续体系形成了凝胶孔径、pH值、缓冲液离子成分的不连续性,这是样品浓缩的主要因素。 蛋白质在聚丙烯酰胺凝胶中电泳时,它的迁移率取决于它所带净电荷以及分子的大小和形状等因素。如果加入一种试剂使电荷因素消除,那电泳迁移率就取决于分子的大小,就可以用电泳技术测定蛋白质的分子量。1967年,Shapiro等发现阴离子去污剂十二烷基硫酸钠(SDS)具有这种作用。当向蛋白质溶液中加入足够量SDS和巯基乙醇,SD S可使蛋白质分子中的二硫键还原。由于十二烷基硫酸根带负电,使各种蛋白质—SDS复合物都带上相同密度的负电荷,它的量大大超过了蛋白质分子原的电荷量,因而掩盖了不同种蛋白质间原有的电荷差别,SDS与蛋白质结合后,还可引起构象改变,蛋白质—SDS复合物形成近似“雪茄烟”形的长椭圆棒,不同蛋白质的SDS复合物的短轴长度都一样,约为1 8A,这样的蛋白质—SDS复合物,在凝胶中的迁移率,不再受蛋白质原的电荷和形状的影响,而取决于分子量的大小由于蛋白质-SDS复合物在单位长度上带有相等的电荷,所以它们以相等的迁移速度从浓缩胶进入分离胶,进入分离胶后,由于聚丙烯酰胺的分子筛作用,小分子的蛋白质可以容易的通过凝胶孔径,阻力小,迁移速度快;大分子蛋白质则受到较大的阻力而被滞后,这样蛋白质在电泳过程中就会根据其各自分子量的大小而被分离。因而S DS聚丙烯酰胺凝胶电泳可以用于测定蛋白质的分子量。当分子量在15KD到200KD之间时,蛋白质的迁移率和分子量的对数呈线性关系,符合下式:logMW=K-bX,式中:MW为分子量,X为迁移率,k、b均为常数,若将已知分子量的标准蛋白质的迁移率对分子量对数作图,可获得一条标准曲线,未知蛋白质在相同条件下进行电泳,根据它的电泳迁移率即可在标准曲线上求得分子量。 SDS-聚丙烯酰胺凝胶电泳经常应用于提纯过程中纯度的检测,纯化的蛋白质通常在SDS电泳上应只有一条带,但如果蛋白质是由不同的亚基组成的,它在电泳中可能会形成分别对应于各个亚基的几条带。SDS-聚丙烯酰胺凝胶电泳具有较高的灵敏度,一般只需要不到微克量级的蛋白质,而且通过电泳还可以同时得到关于分子量的情况,这些信息对于了解未知蛋白及设计提纯过程都是非常重要的。