对硝基苯胺的合成与纯化研究
对硝基苯胺的合成与纯化研究
陆文心(2012301040179)
武汉大学化学与分子科学学院化学弘毅班430072
指导老师:熊英
一、实验目的
1. 以苯胺为原料,经乙酰化、硝化、水解制得对硝基苯胺。
2. 了解乙酰化反应可采取的途径,知道反应机理及反应条件。
3. 认识芳香环上的亲电取代反应,了解其类型和本实验中涉及到的亲电取代反应——硝化反应的机理;理解先将氨基乙酰化的原因,了解苯环上取代基的定位效应。
4. 会配制硝化试剂,理解硝鎓离子产生的机理。
5. 学会用熔点仪测定固体物质的熔点,认识晶体化合物熔点测定的重要性及作用;熟练进行重结晶操作。
6. 认识酰胺在酸催化条件下水解反应的机理,了解本实验中强酸和强碱的作用。
7. 了解薄层层析的原理及操作,并用该方法分析产品的纯度。
8. 了解柱层析的基本原理,掌握柱层析实验的基本操作,用柱层析分离对硝基苯胺和邻硝基苯胺,得到其溶液;初步学会使用旋转蒸发仪,了解旋转蒸发的实质和优点,用该方法获得对硝基苯胺固体。
9. 在老师的指导下对产品对硝基苯胺和邻硝基苯胺进行核磁共振表征,了解化学位移等概念,初步学会分析NMR图谱。
二、实验原理
芳香环上的硝化反应是有机合成中的常见反应,它是一种亲电取代反应。芳香环上常见的亲电取代反应还有卤化、磺化、以及傅-克烷基化和酰基化反应。亲电取代反应由亲电试剂(多为带正电荷的缺电子基团如NO2+等路易斯酸)启动,进攻芳香环上的离域π电子云,将氢原子取代。
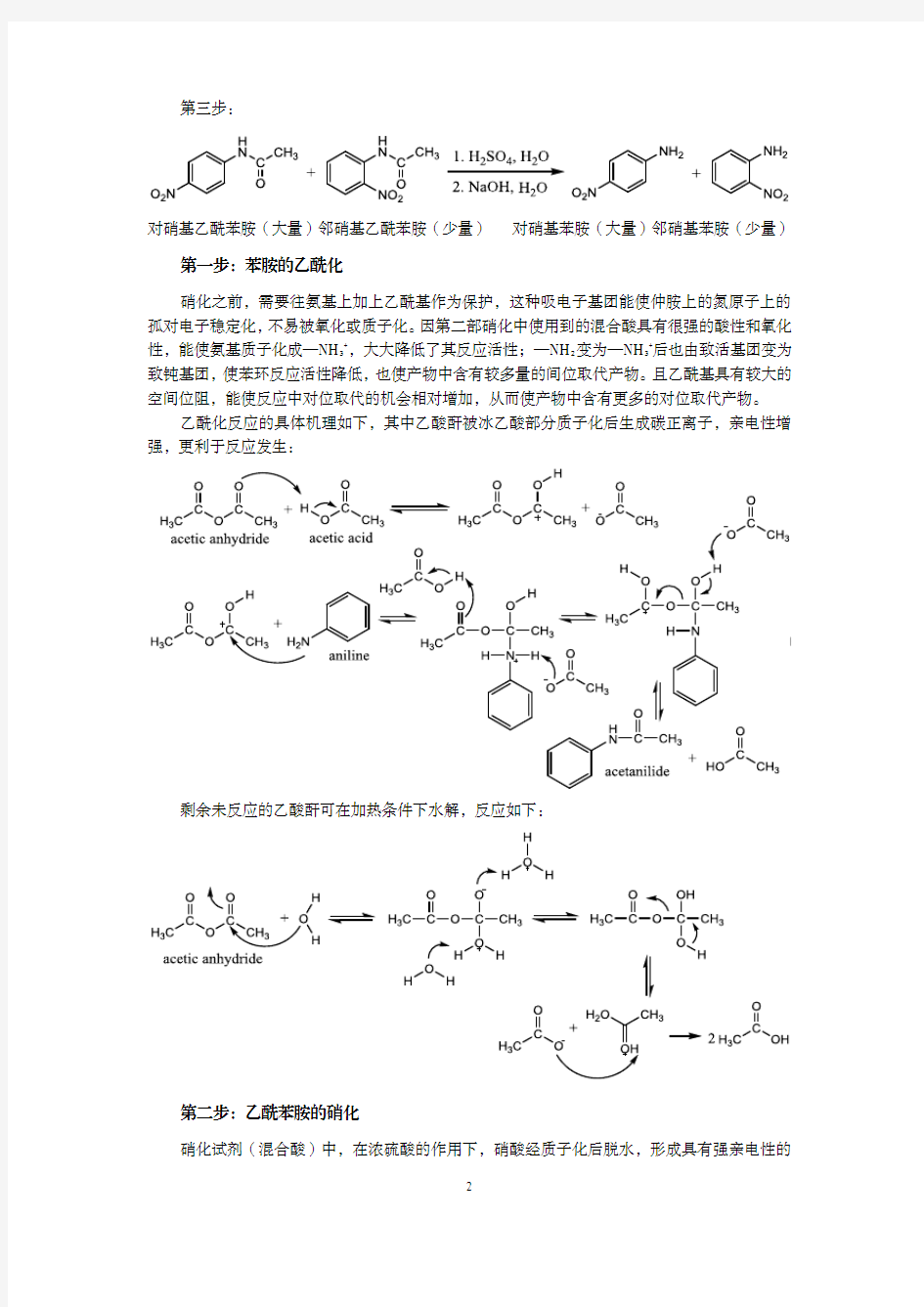
本实验为连续合成实验,分三个主要步骤,从苯胺出发,合成对硝基苯胺。每步的反应式如下:
第一步:
苯胺乙酸酐乙酰苯胺乙酸第二步:
乙酰苯胺对硝基乙酰苯胺(大量)邻硝基乙酰苯胺(少量)
第三步:
对硝基乙酰苯胺(大量)邻硝基乙酰苯胺(少量)对硝基苯胺(大量)邻硝基苯胺(少量)
第一步:苯胺的乙酰化
硝化之前,需要往氨基上加上乙酰基作为保护,这种吸电子基团能使仲胺上的氮原子上的孤对电子稳定化,不易被氧化或质子化。因第二部硝化中使用到的混合酸具有很强的酸性和氧化性,能使氨基质子化成—NH3+,大大降低了其反应活性;—NH2变为—NH3+后也由致活基团变为致钝基团,使苯环反应活性降低,也使产物中含有较多量的间位取代产物。且乙酰基具有较大的空间位阻,能使反应中对位取代的机会相对增加,从而使产物中含有更多的对位取代产物。
乙酰化反应的具体机理如下,其中乙酸酐被冰乙酸部分质子化后生成碳正离子,亲电性增强,更利于反应发生:
剩余未反应的乙酸酐可在加热条件下水解,反应如下:
第二步:乙酰苯胺的硝化
硝化试剂(混合酸)中,在浓硫酸的作用下,硝酸经质子化后脱水,形成具有强亲电性的
硝鎓离子NO2+,进攻苯环,于邻对位(主要在对位)发生硝化反应:
第三步:对硝基乙酰苯胺的水解
酰胺类化合物在强酸和强碱溶液中均能发生水解反应,生成羧酸和铵根离子(酸性水解)或羧酸根离子和氨(碱性水解),反应如下:
在本实验中,将对硝基乙酰苯胺在酸性条件下水解,其反应过程如下:
酸性水解得到的质子化的氨基正离子能溶于水,要得到产品对硝基苯胺,须加入NaOH溶液碱化,反应为:
薄层层析原理
薄层层析(thin layer chromatography),也叫薄层色谱、薄板色谱或薄板层析,属层析技术中的一类,常用TLC代表。其原理与柱层析类似,将粒度细小(通常为200目)的吸附剂(硅胶或氧化铝)均匀涂布于玻璃板、塑料板或金属箔上形成一定厚度的薄层,此为固定相。被分离鉴定的样品制成溶液,用毛细管点在薄层上靠近某一端处,则样品中各种物质都会受到吸附剂或强或弱的吸附而附着在表面。将薄板样品点下端浸入展开剂(某一种溶剂或混合溶剂,作为流动相)中,溶剂靠毛细作用从点有样品的一端向另一端运动并带动样品点前进,各组分分子即在展开剂中发生溶解竞争。经过反复多次的吸附和溶解竞争后,受吸附力较弱而溶解度较大的组分将行进较长的路程;反之受吸附较强或溶解度较小的组分行进的距离则较短,从而使各组分间在板上拉开距离。
薄层层析主要应用于化合物的鉴定和其它分离手段的效果检测,并可监测反应进程、作为柱层析的先导,它作为常用分离方法之一,具有微量、快速、操作简便等优点,通常可分离的量在0.5g以下,最低可达10-9g。
作为检测手段,其理论依据为同种分子在记性、溶解度、分子大小和形状等方面完全相同,因而在同一块板上随展开剂爬升的高度亦应相同;不同种分子在这些方面总会有一些差别,因而其爬升高度不会完全相同。若用其它分离手段所得的某一个组分在薄层板上经样点展开后仍为一个点,则说明该组分为同种分子,即原来的分离方法达到了预期效果;如果展开后变成了几个斑点,则说明该组分中仍有数种分子,即原分离手段未达到预期效果。
作为化合物鉴定的手段,其理论依据是每种化合物都有自己特定的比移值(R f)。比移值的定义为:
R f=化合物样点移动的距离展开剂前沿移动的距离
影响R f值的因素很多,如薄层厚度,吸附剂的种类、粒度、活性,展开剂的纯度(或配比的准确度)及外界温度等。因此,同一化合物在板上重现R f值比较困难,不能仅凭R f值作判断。在鉴定未知样品时用已知化合物在同一块板上点样做对照才比较可靠。
本实验中以涂布于玻璃板上的二氧化硅为固定相,石油醚和乙酸乙酯混合溶剂(体积比3∶1)为流动相,分离对硝基苯胺和邻硝基苯胺,并检验重结晶的分离效果。
柱层析原理
本实验中使用的柱层析属于吸附柱层析。将吸附剂均匀致密得装填在层析柱中,使其呈一圆柱状,为固定相;将待分离的样品制成溶液,从柱顶加入,混合物各组分即吸附在吸附剂颗粒表面。选取合适的溶剂自柱顶向下均匀淋洗,对样品中的各组分分子,溶解度大的易分配进入流动相,而极性小者易被其它分子替换。同种分子各方面相同,下行速率也应相同,不同分子下行思速率不同,即在柱上拉开距离。用不同的接受容器分别接收各组分的溶液,蒸除溶剂即可得到纯品。
旋转蒸发原理
旋转蒸发实质上属于减压蒸馏。通过电动机械控制,烧瓶在最合适的速度下恒速旋转,液体样品在瓶内表面形成一层液体薄膜,增大了蒸发面积。真空泵使烧瓶内处于负压状态。烧瓶在旋转的同时置于水浴锅中恒温加热,瓶内溶液在负压下在烧瓶中进行加热扩散蒸发,蒸气通过双层蛇形冷凝管充分冷却,落入收集瓶。取下烧瓶即可从内壁刮出产品。
核磁共振(氢谱)原理
原子核是带正电的粒子,不能自旋的核没有磁矩,能自旋的核有循环的电流,会产生磁场,
形成磁矩。当自旋核处于某一外加磁场中,两不同自旋的原子核的能量具有一定差异,且磁场强度越强,差异越大。
使用某一频率的光入射,当光子能量与两自旋情况的原子核能量之差相同时,电磁波就会被吸收,以此来检测两能态的能量差值。而由于分子内电子屏蔽的作用,不同化学环境的氢原子自旋基态与激发态能量差值不同,在固定的照射频率时,不同的氢原子核会在不同的磁场强度下显示吸收峰,即不同化学环境的氢原子化学位移不同。根据吸收峰的化学位移值来推断该峰对应的氢原子的化学环境,从而判断待测样品中官能团的种类;通过积分计算不同峰的面积来计算不同氢原子的数量比,从而推断出待测分子具体的结构。
三、主要有机化学品的理化常数
表1 主要有机化学品的理化常数
四、主要化学试剂及仪器
苯胺(A.R.),冰乙酸(A.R.),乙酸酐(A.R.),活性炭,硫酸(A.R.),硝酸(A.R.),乙醇95%(A.R.),氢氧化钠(A.R.),丙酮(A.R.),邻硝基苯胺(C.P.),对硝基苯胺(A.R.),石油醚(60 ~ 90℃,A.R.),乙酸乙酯(A.R.)。
圆底烧瓶(50mL/14#、25mL/14#),锥形瓶(50mL/14#、25mL/14#),球形冷凝管(14#×2),空气冷凝管(14#×2),空心塞(14#),抽滤瓶(150mL),布氏漏斗,三角漏斗,烧杯(250mL、100mL、50mL),量筒(50mL、10mL),温度计(150℃),试管,滴管,表面皿,玻璃棒,pH试纸,层析板(8cm×2.5cm),展开槽,层析柱(2cm×35cm),封口熔点毛细管,点样毛细管,不锈钢铲,搅拌磁子,铁夹,水浴锅,台秤,直尺。
恒温磁力加热搅拌器,加热套,RY-1熔点仪(编号08000236),红外灯,RE-52AA旋转蒸发仪。
五、实验装置图
图1 回流装置图2 抽滤(趁热过滤)装置
六、实验内容(实验现象用楷体注明在括号中)
1. 苯胺的乙酰化
量取5.0mL苯胺,加入50mL圆底烧瓶中,加入10mL冰乙酸将其溶解。在不断搅拌下,逐滴加入6mL乙酸酐。放入磁子,搭建回流装置,缓缓加热回流15min。(加入反应原料以后反应液呈浅黄色,加热后颜色加深,变为红色;回流时冷凝管第一球处出现回流圈,回流接近结束时反应液颜色稍变浅,最终颜色为橙色)
停止加热,移去热源,使装置缓慢冷却。从冷凝管上端管口小心加入5mL冷水,再次煮沸5min。
停止加热,缓缓冷却,将反应液慢慢倒入30mL冰水中并不断搅拌(产生淡黄色沉淀)。在冰水浴中冷却15min,抽滤,获得沉淀,用冰水洗涤沉淀,尽量抽干。留一小匙固体在干净的小烧杯中,待其自然干燥,用熔点仪测定其熔点(数据如下表)。
表2 乙酰苯胺粗品的熔点测定
剩余固体转移到250mL烧杯中,加入100mL水,加热煮沸,搅拌加速溶解。待液体澄清,冷却1min,加入一小匙活性炭,充分搅拌,再次煮沸2min。同时将布氏漏斗放在烘箱中预热,准备热过滤。
取出漏斗,迅速搭建抽滤装置,用几滴沸水润湿滤纸,开泵抽气使滤纸紧贴。倒入正沸腾的溶液,同时开启真空泵抽滤。如有晶体在漏斗中析出,用少量沸水洗掉。
关闭真空泵,取下抽滤瓶在加热器上加热,使滤液澄清,迅速转移入250mL烧杯,盖上表面皿,在室温中缓慢冷却,观察晶体的形成。
冷却至室温后,抽滤得晶体,用真空塞压紧,尽量抽干。转移至表面皿,待其自然干燥,称重(3. 30g),测定熔点(数据如下表)。
表3 重结晶乙酰苯胺的熔点测定
2. 乙酰苯胺的硝化
称取2.4g上一步骤制备的乙酰苯胺,加入50mL锥形瓶中,加入4mL冰乙酸,稍加热溶解(颜色稍变深)。待乙酰苯胺完全溶解后,将锥形瓶放在冰水浴中冷却。逐滴滴入5mL预先冷却的浓硫酸,继续冷却至0℃。
另取一25mL的锥形瓶,加入2mL冰浓硫酸。将锥形瓶浸在冰水浴中,逐滴缓缓加入1.5mL 浓硝酸,冷却10min。(刚加入浓硫酸并冷却时有白色固体出现,加完浓硫酸过一段时间后白色固体消失,液体再次变得透明)
取出盛有乙酰苯胺和硫酸的锥形瓶,在不断搅拌下,以每秒1 ~ 2滴的速率加入刚制备的硝化试剂。插入温度计监测温度,一旦体系温度超过25℃,立即放入冰水浴中冷却(反应中液体呈浅褐色,接近反应结束后变黄)。
继续搅拌,让混合液在室温下放置30min,使硝化反应进行完全。放置时间不可超过1h,以防产物分解或生成二硝基化合物。
量取40mL水,加入约10g冰,在不断搅拌下将反应液缓缓倾入其中(刚加入时出现浅黄绿色沉淀,而后液体变得黏稠)。抽滤,获得产品,反复洗涤沉淀。在沉淀上垫两层滤纸,用真空塞压实,尽量抽干,转移入表面皿中,在红外灯下烘烤,待实验结束放入实验柜,自然干燥。
在试管中装少量粗品,加入2mL 95%乙醇,在水浴中加热溶解后,在室温下缓慢冷却。抽滤得沉淀,转移到小滤纸上,待其自然干燥,测定熔点(数据如下表)。
表4 重结晶对硝基乙酰苯胺的熔点测定
3. 对硝基乙酰苯胺的水解及薄层层析鉴定
称量已干燥的对硝基乙酰苯胺粗品的质量(3.67g),保留少量样品以测定其熔点(数据如下表)。
表5 对硝基乙酰苯胺粗品的熔点测定
剩余产品转移入50mL的圆底烧瓶,加入10mL 1∶1(体积比)硫酸溶液,放入磁子,开动搅拌,加热回流20min。(反应体系开始时非常黏稠,难以搅动,随着温度提高逐渐溶解,最终呈橙黄色)
移去热源,向反应液中加入15mL水,将其尽量完全转移到100mL烧杯中,冷却至室温。逐滴加入5mol·L-1 NaOH溶液(消耗NaOH溶液44mL)至溶液呈强碱性,使胺类沉淀。
抽滤,获得沉淀,用冷水洗涤沉淀,真空塞压实,尽量抽干。
称量湿产品的质量(2.70g),取0.6g产品,放入25mL圆底烧瓶,再加入5mL 75%乙醇,加入磁子,加热回流至固体完全溶解。使溶液冷却至室温,放入冰水浴中冷却15min,观察晶体的析出和生长(析出橙黄色针状结晶,质量为0.22g),抽滤。
取少量粗产品和重结晶后的产品,分别溶解在1mL丙酮中。
取一块8cm×2.5cm的层析板,在距一端沿长度1cm处,用铅笔画一条基线,在线上等距离点两个点(记为1、2)。用毛细管取0.1%邻硝基苯胺溶液、0.1%对硝基苯胺溶液,点在记号点上,要求样点尽量小。在距另一端1cm处也画一条线。
记号点序号与样品溶液的对应见下表。
表6 薄层层析实验中编号与样品对应表
取一个合适的层析缸,要求层析板能够稳定地斜放在其中且盖子仍能盖上。向其中加入3mL 展开剂,盖上盖子几分钟,使蒸气饱和。斜放入层析板,注意液面不得没过样点。盖好盖子,展开剂在毛细作用下上移并带动样点移动。待溶剂前沿到达上部直线时,迅速取出板,令板上的溶剂挥发干,圈出样点最终的位置,并在其中心点上点。
用直尺量出两条线之间的距离,即为S0值;量出样点中心距底部基线的距离,即为S p值。
由公式R f=S p
计算出各样品的比移值如下。
S0
表7 各编号所代表样品的比移值
剩余产品转移至表面皿中,待其自然干燥,供柱层析实验使用。
4. 柱层析法分离对硝基苯胺和邻硝基苯胺
称取0.4g对硝基苯胺,准备进行以下操作。
a. 湿法装柱
取一根层析柱和一根长玻璃棒(长度大于层析柱的深度,一端带有橡皮套)。在层析柱的活塞处涂上真空脂。将层析柱固定在铁夹上,使其呈竖直状态,关闭活塞。通过漏斗向柱中加入少量溶剂(石油醚和乙酸乙酯V/V = 3),观察活塞处是否漏液;在管口下端放一锥形瓶,开启活塞,观察液体能否顺利流下。否则需检查真空脂是否涂得足够,或是否有真空脂堵塞活塞上的小孔。
关闭活塞,向柱中加入一半溶剂,放入一小片棉花,用玻璃棒将其压入底部并挤出气泡。将活塞开到最大,确保液体能以每秒不少于1滴的速率流出。
向一小烧杯中加入20g硅胶,加入少量溶剂,搅拌成悬浮液,迅速加入柱中。打开活塞,用锥形瓶承接液体,继续加入悬浮液,用少量溶剂洗涤烧杯和漏斗内壁,使所有硅胶进入柱中,并使硅胶顶部仍留有1 ~ 2cm厚的液层,用玻璃棒一端的橡皮轻敲柱外壁,使硅胶密实。
b. 干法装样
将0.4g已干燥的对硝基苯胺溶解在约1mL丙酮中,加入1g硅胶,迅速在通风柜中不断搅拌至丙酮完全挥发,形成黄色粉末。
打开活塞使溶剂缓慢流出,至液面距吸附剂顶端约1cm,将粉末加入柱中,轻敲外壁使其平整紧实。用极少量溶剂洗掉管壁上粘附的粉末,放出溶剂至液面接近吸附剂。继续润洗、放出溶剂,重复操作至吸附剂上端的液体呈无色。加入1 ~ 2mm厚的海砂,加入溶剂至接近柱顶。
c. 洗脱、收集
取30支试管,依次编号。打开活塞使液体流下,开始放出的液体呈无色,可不用试管收集,待柱中的黄色部分移动到接近柱底时,开始用试管承接淋洗液,每支试管承接液体的体积可不固定,但须依据柱中的颜色判断是否更换试管。收集时须时刻保持吸附剂上端溶剂的量,保持其始终高于吸附剂,以免气泡被带入吸附剂中。(随着溶剂不断从吸附剂上端通过吸附剂,流出吸附柱,柱内逐渐形成两层黄色色带,黄色色带中间颜色较浅,且色带会随溶剂下移)
d. TLC分析
选取几支试管,用TLC分析鉴定淋洗液,判断哪些试管内的淋洗液为对硝基苯胺溶液、哪些试管内的淋洗液为邻硝基苯胺溶液,合并这些溶液,待下一步蒸除溶剂。薄层层析鉴定的结果如下(S0 = 7.5cm)。
表8 各编号所代表样品的比移值
e. 简单蒸馏与旋转蒸发
由上一步实验中TLC分析结果来看,试管编号为4 ~ 8号的溶液中主要含邻硝基苯胺,试管编号为12号以后的溶液中则更多为对硝基苯胺。
合并样品3 ~ 9号,进行简单蒸馏操作,获得邻硝基苯胺晶体(橙黄色结晶,0.04g);合并样品12 ~ 26号,进行旋转蒸发操作,获得对硝基苯胺晶体(黄色结晶,0.28g)。
取少量上述两种晶体,分别溶解在丙酮中制成稀溶液,进行TLC分析,结果如下。
表9 产品的TLC分析(S0 = 6.5cm)
分别对上述获得的产品进行熔点测定,结果如下。
表10 邻硝基苯胺的熔点测定
表11 对硝基苯胺的熔点测定
5. 邻硝基苯胺和对硝基苯胺的核磁共振(NMR)表征
称取40mg对硝基苯胺产品,小心加入专用的核磁样品管中,加入0.5mL氘代DMSO(二甲亚砜),待其溶解后,放入核磁共振仪中进行表征,获得其NMR图谱。
邻硝基苯胺样品由别的小组提供,同样对其进行核磁共振表征,获得NMR图谱如下。
图3 本小组的对硝基苯胺NMR图谱
图4 对硝基苯胺氢原子的化学位移值
图5 提供的邻硝基苯胺NMR图谱
图6 邻硝基苯胺氢原子的化学位移值
七、产率计算
1. 苯胺的乙酰化产率
取5mL(5.1g)苯胺进行反应,乙酸酐过量,理论应得乙酰苯胺的质量为
5.1g×135.17
93.13
=7.40g
实际得乙酰苯胺3.30g,产率为
3.30g
×100%=44.6%
2. 乙酰苯胺的硝化产率
乙酰苯胺经硝化得到邻硝基苯胺和对硝基苯胺的混合物,此处一并计算。从第一步的产品中
取2.4g进行后续操作,故理论上应得硝基乙酰苯胺的质量为
2.40g×180.16
=3.20g
实际得到硝基乙酰苯胺3.67g,产率为
3.67g
3.20g
×100%=114.7%
3. 硝基乙酰苯胺的水解产率
硝基乙酰苯胺水解得到邻硝基苯胺和对硝基苯胺的混合物。将第二步所得所有产品进行反应,理论上将得到硝基苯胺的质量为
3.67g×138.13
180.16
=2.81g
加上用作重结晶的产品,实际得到的硝基苯胺质量为1.56g,产率为
1.56g
2.81g
×100%=55.5%
4. 柱层析分离两种产物的产率
取0.4g硝基苯胺进行柱层析分离,最终经简单蒸馏和旋转蒸发后得到邻硝基苯胺0.04g、对硝基苯胺0.28g,两者质量比为1∶7。
故邻硝基苯胺的产率为
0.04g
×100%=10.0%
对硝基苯胺的产率为
0.28g
0.40g
×100%=70.0%
5. 本实验的最终产率
由于并不是每一步实验都将上一步的产品全部用来进行反应,故最终产率的计算中应考虑取样占原中间体的比例。若认为对硝基苯胺直接由苯胺硝化而得,可计算出对硝基苯胺的理论产量为
5.1g×138.13
93.13
=7.56g
乙酰苯胺的硝化步骤中,在第一步所得的乙酰苯胺3.30g中取2.4g进行反应,得到1.56g硝基乙酰苯胺,此对硝基乙酰苯胺在后续步骤中完全反应;最后一步中,0.4g硝基苯胺中含0.34g 对硝基苯胺,最终得对硝基苯胺0.28g,故对硝基苯胺的实际收量为
1.56g×3.30g
×
0.28g
=1.50g
对硝基苯胺的最终产率为
1.50g
7.56g
×100%=19.9%
八、实验现象和结果的分析与讨论
1. 对异常实验现象的分析与讨论
第一步苯胺的硝化步骤中,向苯胺中加入乙酸和乙酸酐并加热,溶液变为较深的红色。查阅书籍,获知苯胺易被空气氧化成红色对苯醌。由此知此现象产生可能是由于苯胺被氧化造成,且加热后氧化加剧,使得反应体系颜色变深。
2. 对熔点测定结果的分析与讨论
对乙酰苯胺的熔点测定结果显示,重结晶效果不太明显,重结晶前后的物质熔点相同,说明此部分重结晶只是使得乙酰苯胺的晶形更完整,对乙酰苯胺纯度的提高作用不大。但两者的熔点均接近理论值,说明乙酰苯胺的纯度较高,可以直接用于第二步反应。
对对硝基乙酰苯胺的熔点测定结果显示,此重结晶效果显著,重结晶前的样品的熔点只有148℃左右,明显低于理论值,且固体带浅黄绿色,与文献显示的对硝基乙酰苯胺性状有较大偏差,说明重结晶前此产品纯度较低,需要提纯;重结晶后的样品熔点接近理论值,且重结晶后成为近乎白色的固体,与文献所述性状很接近,说明此步重结晶作用明显,达到了提纯的效果。重结晶后的产品熔程扩大,因为目视观察样品是否熔化时存在一定误差。
最终产物的熔点测定结果与性状均与文献值接近,尤其是邻硝基苯胺成橙黄色,很像纯品,结合柱层析后TLC分析结果,同一种物质的样品点的位置基本处于同一水平线,说明柱层析分离效果已达到。
3. 对薄层层析结果的分析与讨论
a. 对硝基乙酰苯胺水解后的TLC分析
板1是标准样品,将板2和板3和板1进行比较,可以很明显地看出,重结晶前的产品样点和重结晶后的产品样点均分为两个点,但重结晶前的样品点颜色上下均较深,重结晶后的样品点颜色上浅下深,由于第一块板上显示,比移值大的为邻硝基苯胺,比移值小的为对硝基苯胺,故认为重结晶起到了分离纯化效果,有效地纯化了对硝基苯胺。
b. 柱层析所得样品的TLC分析
由表8可以看出,2号样品的比移值比邻硝基苯胺的比移值还要大,分析可能是多硝基取代物;前10个样品的比移值均较大,应为邻硝基苯胺;12以后的比移值较小,应为对硝基苯胺。同一种产物的样点基本在同一水平线上,故分离效果较好。
4. 对核磁共振图谱的分析与讨论
将对硝基苯胺作为样品得出图3、图5,而图4、图6是用ChemDraw模拟计算出的邻硝基苯胺和对硝基苯胺分子中不同氢原子的化学位移值。
a. 对硝基苯胺
由图3和图4可看出,对硝基苯胺在化学位移值为6.6 ~ 8的区间内有三个峰,峰位移值分别为8.006、7.980(分裂),6.787,6.660、6.634(分裂),峰面积比约为1∶1∶1。由分子模拟得出的图4对比可知,对硝基苯胺分子中有三种化学环境不同的氢原子,8左右的峰应为硝基邻位上的氢原子,6.787的峰应为氨基上的氢原子,6.6的峰应为氨基邻位上的氢原子(为何不是6.6的峰为氨基氢6.787的峰为氨基邻位氢,后面会作出解释)。且三种氢原子个数比为1∶1∶1。
在3.4附近出现的峰为水分子,在2.5附近出现的峰为氘代DMSO(溶剂的影响),在0附近出现的峰为硅脂,极有可能是实验中活塞处的硅脂溶于溶剂中导致。
b. 邻硝基苯胺
由图5和图6可看出,邻硝基苯胺在化学位移值为6.6 ~ 8的区间内有四个峰,峰面积比约为1∶3∶1∶1。
由分子模拟得出的图5对比可知,邻硝基苯胺分子中应有五种化学环境不同的氢原子,氨基氢为7.42,氨基邻位为6.76,硝基对位为7.57,氨基对位为7.40,硝基邻位为8.09,且三种氢原
子个数比为2∶1∶1∶1∶1。造成理论与实际不一致的结果,是氨基氢与氨基对位上的氢原子化学位移值十分接近,发生了重叠,成为一个无法分辨的峰,这两种氢原子个数加起来也正好为3,符合实验结果。故图谱中四个峰从左到右分别对应硝基邻位、氨基和氨基对位、硝基对位、氨基邻位。
c. 经重水交换的对硝基苯胺
向一份对硝基苯胺样品中加入重水进行氢交换,得到了如下NMR图谱。
图7 重水氘代的对硝基苯胺NMR图谱
与图3对比可以发现,化学位移值为8的峰和6.6左右的峰仍然存在,且比例仍约为1∶1,说明这两个峰对应的是苯环上的氢的峰,与经分子模拟出的图4对比可知,8.0左右峰对应硝基邻位,6.6左右的峰对应氨基邻位。由图7可知,加入重水进行氢交换之后,原本在6.787处的一个峰消失,又由于氨基上的氢受苯环的影响变得活泼,故认为说明原在6.787的峰对应的是氨基氢,加入重水后,这两个活泼氢被氘取代,因此原有的峰消失。同样,原本属于水的在3.4左右的峰也消失了,说明重水的加入也会替换掉水中的氢原子。此外,在4.4附近产生了一个新的峰,此峰应为重水的峰。
5. 对产率的分析与讨论
这几个步骤中,苯胺乙酰化的产率较低,分析可能原因如下:①对乙酰苯胺粗品进行了重结晶,而重结晶过程热过滤会损失相当一部分的乙酰苯胺(在滤纸、布氏漏斗等处),且热过滤的滤液为乙酰苯胺的饱和溶液,无法回收出乙酰苯胺,此原因应该是此步骤产率较低的主要原因;
②苯胺在烧瓶中反应后反应液黏度较大,不能够完全转移到烧杯中;③抽滤完毕以后滤纸上固体的转移不充分。
乙酰苯胺的硝化步骤中,产率超过了100%,结合多数同学的结果,发现较多同学这一步的产率都超过了100%,分析其原因,可能有如下几点:①此步骤所得的硝基乙酰苯胺固体极为黏
稠,粘在一起成为块状,不仅不易在布氏漏斗中压实(会大量黏附于空心塞上),也不易在室温中干燥,黏附于空心塞上的产品不易回收,成为损失,产品不易在室温中干燥使得称重时其中仍含有部分水分,这也是造成产率虚高的原因。
最终产率约为20%。造成最终产率较低有下述原因:①在所有固体转移的操作中,都不能保证所有固体完全转移到目标容器中,有部分固体洒落,也有少量固体会残留在表面皿或称量纸上,最后旋转蒸发时,烧瓶中的对硝基苯胺也不能完全刮出;②硝基乙酰苯胺和硝基苯胺均有一定的挥发性,实验中发现,若把盛有产品的表面皿放在实验柜中,暴露在空气中的产品会挥发而使得周围的柜壁变黄,说明产品具有挥发性,挥发也使得实验中间体质量减少,也使得最终产率降低;③每制备出一种中间体,均需测量其熔点,虽然取样很少,但积少成多,取出的用于测定熔点的物质也使得最重的目标产品质量有轻微的减少。
对硝基苯胺的制备及纯化
对硝基苯胺的制备 段东斑 (武汉大学化学与分子科学学院湖北武汉430072)
目录 一、实验目的-------------------------------------------------------3 二、实验原理-------------------------------------------------------3 2.1合成-----------------------------------------------------------3 2.2产品的分离与纯化-------------------------------------------4 三、主要试剂及产物的物理常数--------------------------------5 四、主要试剂规格、用量-----------------------------------------6 五、实验装置图-----------------------------------------------------6 六、实验步骤与现象-----------------------------------------------6 6.1苯胺的乙酰化--------------------------------------------------7 6.2乙酰苯胺的硝化---------------------------------------------7 6.3硝基乙酰苯胺的水解-----------------------------------------7 6.4柱层析与薄层层析------------------------------------------8 6.5蒸馏-----------------------------------------------------------8 七、产品的表征与纯度分析-------------------------------------9 7.1熔点的测定--------------------------------------------------9 7.2薄层色谱(TLC)---------------------------------------------10 7.3核磁共振氢谱1HNMR -------------------------------------10 八、产率计算及分析---------------------------------------------11 九、讨论------------------------------------------------------------12 十、其他合成方法------------------------------------------------13
对硝基苯胺的制备
对硝基苯胺的制备(一) 一、实验目的 了解芳香族硝基化合物的制备方法,尤其是由芳胺制备芳香族硝基化合物的方法。 二、实验原理 主反应: +CH 3COOH O 2N NH 2H 2SO 4H 2SO 4H 2O +O 2N NHCOCH 3H 2O ++O 2N NHCOCH 3HONO 2 NHCOCH 3 副反应: +CH 3COOH NHCOCH 3NO 2H 2SO 4H 2SO 4 H 2O +NHCOCH 3H 2O ++NH 2HONO 2 NHCOCH 3 三、实验药品 乙酰苯胺5g (),硝酸(d =)(),浓硫酸,冰醋酸,乙醇,碳酸钠,20%氢氧化钠溶液。 四、实验仪器 锥形瓶,烧杯,滴管,抽滤装置。 五、实验步骤 1.对硝基乙酰苯胺的制备 在100ml 锥形瓶内,放入5g 乙酰苯胺和5mL 冰醋酸[1] 。用冷水冷却, 一边摇动锥形瓶,一边慢慢地加入10mL 浓硫酸。乙酰苯胺逐渐溶 解。将所得溶液放在冰盐浴中冷却到0~2℃。 在冰盐浴中用浓硝酸和 浓硫酸配置混酸。一边摇动锥形瓶,一边 用吸管慢慢地滴加此混酸,保持反应温度不超过5℃[2]。 从冰盐浴中取出锥形瓶,在室温下放置30min ,间歇摇荡之。在搅 拌下把反应混合物以细流慢慢地倒入20mL 水和20g 碎冰的混合物中,对硝基乙酰苯胺立刻
成固体析出。放置约10min,减压过滤,尽量挤压掉粗产品中的酸液,用冰水洗涤三次,每次用10mL。称取粗产品(样品A),放在空气中晾干。其余部分[3]用95%乙醇进行重结晶[4]。减压过滤从乙醇中析出的对硝基乙酰苯胺,用少许冷乙醇洗涤,尽量压挤去乙醇。将得到的对硝基乙酰苯胺(样品B)放在空气中晾干。 将所得乙醇母液在水浴上蒸发到其原体积的2/3。如有不溶物,减压过滤。保存母液(样品C)。 2.对硝基乙酰苯胺的酸性水解 在50mL圆底烧瓶中放入4g对硝基乙酰苯胺和20mL 70%硫酸[5],投入沸石,装上回流冷凝管(如图),加热回流10-20min[6]。将透明的热溶液倒入100mL冷水中。加入过量的20%氢氧化钠溶液,使对硝基苯胺沉淀下来。冷却后减压过滤。滤饼用冷水洗去碱液后,在水中进行重结晶[7]。 纯对硝基苯胺为黄色针状晶体,熔点℃。 附注 [1] 乙酰苯胺可以在低温下溶解于浓硫酸里,但速度较慢,加入冰醋酸可加速其溶解。 [2] 乙酰苯胺与混酸在5℃下作用,主要产物是对硝基乙酰苯胺;在40℃作用,则生成约25%的邻硝基乙酰苯胺。 [3] 也可用下法除去粗产物中的邻硝基苯胺。将粗产物放入一个盛20 mL水的锥形瓶中,在不断搅拌下分次加入碳酸钠粉末,直到混合液对酚酞试纸显碱性。将反应混合物加热至沸腾,这时对硝基乙酰苯胺不水解,而邻硝基乙酰苯胺则水解为邻硝基苯胺。混合物冷却到50℃时,迅速减压过滤,尽量挤压掉溶于碱液中的邻硝基苯胺,再用水洗涤并挤压去水分。取出晾干。 [4] 利用邻硝基乙酰苯胺和对硝基乙酰苯胺在乙醇中溶解度的不同,在乙醇中进行重结晶,可除去溶解度较大的邻硝基乙酰苯胺。 [5] 70%硫酸的配制方法:在搅拌下把4份(体积)浓硫酸小心地以细流加到3份(体积)冷水中。 [6] 可取1mL反应液加到2~3mL水中,如溶液仍清澈透明,表示水解反应已完全。
对硝基苯胺的合成
对-硝基苯胺的制备MSDS 化合物名称 分子 量 性状 比重 (d ) 熔点 (℃) 沸点 (℃) 折光率 (n) 溶解度 水乙醇乙醚 苯胺93.12 液体 1.022 -6.1 184.4 1.586 3.618∞∞冰醋酸60.05 液体 1.049 16.5 118.1 1.371 ∞∞∞ 乙酰苯胺135 .1 斜方晶 体 1.214 133.4 305 - 0.53 3.580 21.20 46.60 7.25 对硝基苯胺138 .1 淡黄色 针状结 晶 1.424 148.5 331.7 0.000 8 邻硝基苯胺138. 12 橙黄色 针状结 晶 1.44 69.7 284.5 一、实验目的 1利用乙酰苯胺制备对-硝基苯胺; 2掌握连续合成的方法,复习抽滤、重结晶等实验基础操作。 二、实验原理 由于氨基对于苯环是强活化基团(亲电试剂主要进攻其邻对位),故可生成对硝基苯胺及邻硝基苯胺,降低了对-硝基苯胺的产率,因此我们用乙酰基对氨基进行保护。而且,加入乙酰基后,由于其空间结构较大并且降低了氨基在亲电取代反应(特别是卤化)中的活化能力,使氨基由很强的第1类定位基变成中等强度的第1类定位基,使反应由多元取代变为有用的一元取代,这些均有利于后来的硝基在对位进行取代。在某些情况下,酰化可以避免氨基与其它功能基或试剂(如RCOCl,-SO2Cl,HNO2等)之间发生不必要的反应。 综合以上考虑,本实验中采用“乙酰苯胺→对-硝基乙酰苯胺→对-硝基苯胺”步骤进行合成。 以乙酰苯胺为反应物制备对-硝基乙酰苯胺,进而脱保护制备对-硝基苯胺,反应方程式如下:
NHCOCH3 +HNO3H2SO4 HOAc NHCOCH3NHCOCH3 + NO2 NO2 NHCOCH3NHCOCH3 + NO2 NO2NH2NH2 + NO2 NO2 +H2O KOH EtOH +CH3COOK 在制备对-硝基乙酰苯胺时,用醋酸做溶剂同时可以防止乙酰苯胺或对-硝基乙酰苯胺水解。对于产物来说,酸或碱都能够促使其水解,为了将粗产物中残留的酸中和掉而又不过量,实验中使用磷酸二氢钠,其中和结果是一种pH接近中性的缓冲溶液。由于邻-硝基苯胺形成分子内氢键,沸点低,对-硝基苯胺形成分子间氢键,沸点高,所以二者可以用水蒸气蒸馏的方法进行分离。 三、实验试剂 苯胺、浓盐酸、活性炭、乙酸酐、乙酸钠、冰醋酸、浓硫酸、浓硝酸、冰块、15%磷酸氢二钠溶液、95%乙醇、1:1硫酸、20%氢氧化钠溶液、石油醚、丙酮 四、实验步骤 乙酰苯胺→对-硝基乙酰苯胺→对-硝基苯胺 1..对-硝基乙酰苯胺的制备 在干燥的50ml锥形瓶中放入5克乙酰苯胺(0.037mol),加入7.9ml冰醋酸,加热至溶解。稍冷后相继用冷水浴和冰水浴冷却到10℃,滴入7.9ml浓硫酸,再在冰水浴中冷到10℃左右,溶液变得浓稠。 在干燥的25ml锥形瓶中混合3ml浓硝酸(含硝酸约0.044mol)和2.1ml浓硫酸,塞住瓶口,用冷水浴冷却到10℃到15℃,然后用滴管慢慢滴加到已制备的乙酰苯胺溶液中,边滴加边摇匀,控制反应温度在15℃到20℃之间,10到15分钟滴完。之后再室温下放置半小时以上,并注意监视温度变化。若发现温度上升超过室温,应以冰水浴冷却到15℃,然后重新在室温下放置并观察温度变化,直至在室温下连续放置半小时而温度不超过室温为止。 在100ml烧杯中放置42.5ml水和10克碎冰,将反应混合物倾注其中,搅拌,抽滤,尽量压干。 将固体转移到100ml烧杯中,加入15%磷酸氢二钠水溶液42.5到45ml,使液体呈中性,搅拌成糊状,抽滤。用约15ml水荡洗烧杯,一并转入抽滤漏斗,抽干后再用约25ml冷水洗涤滤饼,重新抽滤,用玻璃塞挤压滤饼,尽量抽干。将固体转移到表玻璃上晾干。分出一半产物,用95%乙醇重结晶纯化,另外一半不作处理,分别编号为A和B。 2.对-硝基乙酰苯胺的纯度测定 取少许A和B的晶体做熔点测定,记录三次熔点测定数值。 以石油醚与丙酮的等体积混合液作为展开剂进行薄层层析,计算比移值与样点数。
最新对硝基苯胺的制备
对硝基苯胺的制备(一) 一、实验目的 了解芳香族硝基化合物的制备方法,尤其是由芳胺制备芳香族硝基化合物的方法。 二、实验原理 主反应: +CH 3COOH O 2N NH 2H 2SO 4H 2SO 4H 2O +O 2N NHCOCH 3H 2O ++O 2N NHCOCH 3HONO 2 NHCOCH 3 副反应: +CH 3COOH NHCOCH 3NO 2H 2SO 4H 2SO 4 H 2O +NHCOCH 3H 2O ++NH 2HONO 2 NHCOCH 3 三、实验药品 乙酰苯胺5g (0.037mol ),硝酸(d =1.40)2.2mL (0.032mol ),浓硫酸,冰醋酸,乙醇,碳酸钠,20%氢氧化钠溶液。 四、实验仪器 锥形瓶,烧杯,滴管,抽滤装置。 五、实验步骤 1.对硝基乙酰苯胺的制备 在100ml 锥形瓶内,放入5g 乙酰苯胺和5mL 冰醋酸[1]。用冷水冷 却,一边摇动锥形瓶,一边慢慢地加入10mL 浓硫酸。乙酰苯胺逐 渐溶解。将所得溶液放在冰盐浴中冷却到0~2℃。 在冰盐浴中用2.2mL 浓硝酸和1.4mL 浓硫酸配置混酸。一边摇动 锥形瓶,一边用吸管慢慢地滴加此混酸,保持反应温度不超过5℃ [2]。 从冰盐浴中取出锥形瓶,在室温下放置30min ,间歇摇荡之。在搅 拌下把反应混合物以细流慢慢地倒入20mL 水和20g 碎冰的混合物 中,对硝基乙酰苯胺立刻成固体析出。放置约10min ,减压过滤, 尽量挤压掉粗产品中的酸液,用冰水洗涤三次,每次用10mL 。称 取粗产品0.2g (样品A ),放在空气中晾干。其余部分[3]用95%乙醇进行重结晶[4]。减压过滤从乙醇中析出的对硝基乙酰苯胺,用少许冷乙醇洗涤,尽量压挤去乙醇。将得到的对硝基乙酰苯胺(样品B )放在空气中晾干。 将所得乙醇母液在水浴上蒸发到其原体积的2/3。如有不溶物,减压过滤。保存母液(样品
对硝基苯胺的制备.doc1111
对硝基苯胺的合成实验 一.对硝基苯胺的基本理化性质 淡黄色针状结晶,易于升华。 熔点148.5℃, 沸点331.7 ℃, 相对密度1.424(20/4℃)。 闪点199°F[1], 水中溶解度为0.0008g。微溶于冷水,溶于沸水、乙醇、乙醚、苯和酸溶液。 该品有毒,空气中容许浓度为5mg/m3。吸入、口服和皮肤接触有害。 毒性高毒。可引起比苯胺更强的血液中毒。如果同时存在有机溶剂或在饮酒后,这种作用更为强烈。急性中毒表现为开始头痛、颜面潮红、呼吸急促,有时伴有恶心、呕吐,之后肌肉无力、发绀、脉搏频弱及呼吸急促。皮肤接触后会引起湿疹及皮炎。 二.预备知识 芳胺的酰化在有机合成中的作用: (1)乙酰化反应常被用来“保护”伯胺和仲胺官能团,以降低芳胺对氧化性试剂的敏感性。 (2)氨基经酰化后,降低了氨基在亲电取代反应(特别是卤化)中的活化能力,使其由很强的第I类定位基变成中等强度的第I类定位,使反应由多元取代变为有用的一元取代。 (3)由于乙酰基的空间效应,往往选择性地生成对位取代产物。(4)在某些情况下,酰化可以避免氨基与其它功能基或试剂(如RCOCl,-SO2Cl,HNO2等)之间发生不必要的反应。
乙酰苯胺可由苯胺与酰氯、酸酐或是冰醋酸来制备,由于是实验室制备,所以选成本较小且污染小的冰醋酸来进行乙酰化,冰醋酸是一种无色液体,有强烈刺激性气味。熔点16 .6℃,沸点117 .9℃,是典型的脂肪酸。被公认为食醋内酸味及刺激性气味的来源。在家庭中,乙酸稀溶液常被用作除垢剂。食品工业方面,在食品添加剂列表E260中,乙酸是规定的一种酸度调节剂。 三.实验原理 1.乙酰苯胺的制备原理 乙酰苯胺为无色晶体,具有退热镇痛作用,是较早使用的解热镇痛药,因此俗称“退热冰”。乙酰苯胺也是磺胺类药物合成中重要的中间体。由于芳环上的氨基易氧化,在有机合成中为了保护氨基,往往先将其乙酰化转化为乙酰苯胺,然后再进行其他反应,最后水解除去乙酰基。 乙酰苯胺可由苯胺与乙酰化试剂如:乙酰氯、乙酐或乙酸等直接作用来制备。反应活性是乙酰氯>乙酐>乙酸。由于乙酰氯和乙酐的价格较贵,本实验选用纯的乙酸(俗称冰醋酸)作为乙酰化试剂。反应式如下: 冰醋酸与苯胺的反应速率较慢,且反应是可逆的,为了提高乙酰苯胺的产率,一般采用冰醋酸过量的方法,同时利用分馏柱将反应中生成的水从平衡中移去。由于苯胺易氧化,加入少量锌粉,防止苯胺在反应过程中氧化。 2.由乙酰苯胺合成对硝基苯胺的原理
以硝基苯为原料合成对溴苯胺
2011级化学教育有机化学 综合性与设计性实验 题目:以硝基苯为原料合成对溴苯胺以硝基苯为原料合成对溴苯胺 (华南师范大学化学与环境学院) 摘要对溴苯胺是非常重要的有机化工原料,常被用作染料原料,如偶氮染料、喹啉染料等,医药及有机合成的中间体等。本实验合成过程以硝基苯为原料,经历制备苯胺、乙酰苯胺、对溴乙酰苯胺等中间体的过程,最终制得目标产物对溴苯胺。其合成过程经历硝化、还原、保护、溴代、去保护等多个步骤,可以制得纯度较高的对溴苯胺。同时,掌握了芳烃硝化、硝基的还原、氨基的保护与去保护、芳烃卤代等方法。通过实验可得,用此实验方法制备对溴苯胺,操作方法简单,可控性强。 关键词合成;对溴苯胺;硝基苯;苯胺;乙酰苯胺;对溴乙酰苯胺Abstract P-bromo-aniline is very important to the organic chemical raw materials, dyes were often used as raw materials, such as the azo dyes, kuilin dyes, medicine and synthetic organic intermediates, etc. The synthesis process of nitrobenzene in as raw material, through preparation aniline, acetyl aniline, bromine acetyl aniline intermediates such as to the process, finally made of bromine aniline target product.
由苯胺设计合成对硝基苯胺
实验名称由苯胺设计合成对硝基苯胺院系化学化工学院 班级化基1101 学号20110903215 姓名刘永超
一、实验目的 1、掌握由苯胺设计合成对硝基苯胺的原理 2、掌握邻硝基苯胺和对硝基苯胺的分离方法 3、学会对有毒药品的操作和处理 二、预备知识 1、反应中各步化合物的物理性质 化合物 名称 分子量性状熔点℃沸点℃溶解度 水乙醇乙醚苯胺93.12 无色油 状液体 -6.3 184 微溶溶溶 乙酸酐102.09 无色透 明液体 -73.1 138.6 微溶溶溶 乙酰苯胺135.16 斜方晶 体 133.4 305 微溶于 冷水, 溶于热 水 溶溶 对硝基乙酰苯胺180.16 无色晶 体 100 215.6 微溶于 冷水, 易溶于 沸水 溶溶 邻硝基乙酰苯胺180.16 淡黄色 片状 94 100 微溶于 冷水, 易溶于 沸水 溶溶 对硝基苯胺138.12 淡黄色 针状 148.5 331.7 微溶于 冷水, 易溶于 沸水 溶溶 邻硝基苯胺138.12 橙黄色 针状 69.7 284.5 微溶于 冷水, 易溶于 沸水 溶溶 2、酰化反应的反应活性:酰氯>酸酐>酯>酰胺,故乙酰苯胺可由苯胺与酰氯、酸酐或冰醋酸来制备。在芳胺的反应中,常将氨基通过乙酰化反应保护起来,降低了氨基在亲电取代反应中的活化能力,以
防止氨基被氧化。由于乙酰基的空间位阻效应,酰胺基属于邻对位定位基,在苯环上往往选择性地生成邻对位取代产物。 三、实验原理 先以苯胺为原料,经乙酰化合成乙酰苯胺,再经过硝化,水解得到邻硝基苯胺和对硝基苯胺的混合物,再通过蒸馏,柱层析,或水蒸气蒸馏分离即可得到对硝基苯胺。 1、乙酰苯胺的制备 乙酸和苯胺的反应是可逆的,且反应速率较慢,可采用乙酸过量的方法和利用分馏柱将反应中生成的水蒸除,使平衡向水生成的方向移动而提高乙酰苯胺的产率。 2、硝化反应 乙酰苯胺与混酸反应,硝化的位置与温度有关。在低温(低于5℃)下产物以对硝基乙酰苯胺为主。硝化温度升高,邻硝基乙酰苯胺产物将增多。 3、水解反应:
对硝基苯胺的合成与纯化研究
对硝基苯胺的合成与纯化研究 陆文心(2012301040179) 武汉大学化学与分子科学学院化学弘毅班430072 指导老师:熊英 一、实验目的 1. 以苯胺为原料,经乙酰化、硝化、水解制得对硝基苯胺。 2. 了解乙酰化反应可采取的途径,知道反应机理及反应条件。 3. 认识芳香环上的亲电取代反应,了解其类型和本实验中涉及到的亲电取代反应——硝化反应的机理;理解先将氨基乙酰化的原因,了解苯环上取代基的定位效应。 4. 会配制硝化试剂,理解硝鎓离子产生的机理。 5. 学会用熔点仪测定固体物质的熔点,认识晶体化合物熔点测定的重要性及作用;熟练进行重结晶操作。 6. 认识酰胺在酸催化条件下水解反应的机理,了解本实验中强酸和强碱的作用。 7. 了解薄层层析的原理及操作,并用该方法分析产品的纯度。 8. 了解柱层析的基本原理,掌握柱层析实验的基本操作,用柱层析分离对硝基苯胺和邻硝基苯胺,得到其溶液;初步学会使用旋转蒸发仪,了解旋转蒸发的实质和优点,用该方法获得对硝基苯胺固体。 9. 在老师的指导下对产品对硝基苯胺和邻硝基苯胺进行核磁共振表征,了解化学位移等概念,初步学会分析NMR图谱。 二、实验原理 芳香环上的硝化反应是有机合成中的常见反应,它是一种亲电取代反应。芳香环上常见的亲电取代反应还有卤化、磺化、以及傅-克烷基化和酰基化反应。亲电取代反应由亲电试剂(多为带正电荷的缺电子基团如NO2+等路易斯酸)启动,进攻芳香环上的离域π电子云,将氢原子取代。 本实验为连续合成实验,分三个主要步骤,从苯胺出发,合成对硝基苯胺。每步的反应式如下: 第一步: 苯胺乙酸酐乙酰苯胺乙酸第二步: 乙酰苯胺对硝基乙酰苯胺(大量)邻硝基乙酰苯胺(少量)
由苯胺合成对硝基苯胺
由苯胺设计合成对硝基苯胺 一、实验目的 1.了解由苯胺和乙酸酐制备对硝基苯胺的原理及方法。 2.了解水蒸气蒸馏,分馏,柱层析分离 3.熟悉固体样本熔点的测定方法 4.掌握重结晶的操作步骤和方法 5.掌握氨基的保护和去保护的原理和实验操作 二、实验原理 芳环上的氨基易被氧化,因此由苯胺制备对硝基苯胺,不能直接硝化,须先保护氨基。 将苯胺转化为乙酰苯胺,保护氨基后再硝化,在芳环引入硝基后,再水解去保护恢复氨基,从而得到对硝基苯胺。另外,氨基酰化后,降低了氨基对苯环亲电取代反应的活化能力,又因为乙酰基的空间效应,可提高生成对位产物的选择性。 1、苯胺的乙酰化 乙酸与苯胺的反应是可逆的,且反应速率较慢,实验中使用过量乙酸,利用分馏柱将反应中生成的水蒸气除去使平衡向右移动而提高乙酰苯胺的产率。 2、对硝基乙酰苯胺的制备乙酰苯胺与混酸反应,硝化的位置与温度有关,低于5℃时产物以对硝基苯胺为主,硝化温度升高,邻硝基苯胺产物增多。
3、除邻位副产物pH=10时,邻位产物较对位产物易水解,生成的邻硝基苯胺又溶于50℃的碱液,故将混合产物与碳酸钠溶液共沸水解,50℃过滤即可除去邻位副产物。对位产物再与氢氧化钠溶液共沸,水解得对硝基苯胺。 三、实验试剂及主要参数
四、实验步骤及现象 1.由乙酰苯胺合成对硝基苯胺
2.薄层层析法检验纯度 取一块已铺好硅胶的薄板( 只能碰触边缘和背面) 和层析缸,向层析缸中加入3ml 展开剂(乙酸乙酯与石油醚1: 3混合物)盖好盖子;在薄板距下边缘约1cm 处用软铅笔画一条水平横线,点三个点。取两端开口的毛细管,在1%的对硝基苯胺丙酮溶液中蘸一下,在第一个点处轻点,如颜色太浅,可待丙酮完全挥发后再点一次。用1%邻硝基苯胺点第二个点,用自己配制的粗对硝基苯胺丙酮溶液点第三个点,然后将薄板小心放入层析缸中,注意边缘不要碰壁。待溶剂线靠近上边缘后,取出薄板,用铅笔划线标记展开剂上升的高度,圈出各点的轮廓并点出中点。 实验结果分析:粗对硝基苯胺丙酮溶液在硅胶板上有两个点,分别与对硝基苯胺和邻硝基苯胺对齐,与邻硝基苯胺对齐的点颜色较淡,说明产物较纯,含杂质较少。 3.水蒸气蒸馏
对硝基苯胺
对硝基苯胺 p-nitroaniline 分子式C6H6N2O2 相对分子质量138.12, CAS NO 100-01-6 名称:4-硝基苯胺 别名:对硝基苯胺 主要性质: 淡黄色针状结晶,易于升华。 熔点148.5℃, 沸点331.7 ℃, 相对密度1.424(20/4℃)。 闪点199°F[1], 水中溶解度为0.0008g。微溶于冷水,溶于沸水、乙醇、乙醚、苯和酸溶液。 该品有毒,空气中容许浓度为5mg/m3。吸入、口服和皮肤接触有害。 毒性高毒。可引起比苯胺更强的血液中毒。如果同时存在有机溶剂或在饮酒后,这种作用更为强烈。急性中毒表现为开始头痛、颜面潮红、呼吸急促,有时伴有恶心、呕吐,之后肌肉无力、发绀、脉搏频弱及呼吸急促。皮肤接触后会引起湿疹及皮炎。大鼠经口LD50为1410mg/kg。 生产方法 工业生产对硝基苯胺。可采用乙酰苯胺硝化、水解的方法,也可用对硝基氯苯氨解的方法。 1.以乙酰苯胺为原料,经硝化、水解而制得。 原料消耗定额:乙酰苯胺1210kg/t;硝酸(90%)580kg/t;硫酸3620kg/t;液碱(30%)660kg/t。 2.以对硝基氯苯为原料,可采用高压釜间歇法生产,也可采用管道反应器连续化生产,收率都在94%左右。 原料消耗定额:对硝基氯苯(97%)1170kg/t;氨水(28%)700kg/t。 产品用途 对硝基苯胺是染料工业极为重要的中间体,可直接用于合成品种有:直接耐晒黑 G,直接绿B、BE、2B-2N,黑绿NB,直接灰D、酸性黑10B、ATT,分散红P-4G、阳离深黄2RL,毛皮黑D,对苯二胺,邻氯对硝基苯胺,
2.6-二氯-4硝基苯胺,5-硝基-2-氯苯酚等,也可合成农药氯硝胺,医药卡柳肿;同时还是防老剂,光稳定剂,显影剂等的原料。国外以对硝基苯胺为重氮组份合成的分散染料有:C,I分散橙1,3,7,21等、红色1,2,7,17等,蓝259;黑2,3,28,29等 该品即冰染染料大红 GG色基,可作黑色盐 K,供棉麻织物染色、印花之用。但主要用作偶氮染料中间体,如用于生产直接墨绿B、酸性媒介棕G、酸性黑10B、酸性毛元ATT、毛皮黑D和直接灰D等。还可作农药和兽药的中间体,在医药工业中可用于生产氯硝柳胺、卡巴肿、硝基安定、喹啉脲硫酸盐等。还可用于生产对苯二胺;抗氧化剂和防腐剂等。 应急处理 隔离泄漏污染区,限制出入。切断火源。建议应急处理人员戴防尘面具(全面罩),穿防毒服。不要直接接触泄漏物。小量泄漏:避免扬尘,用洁净的铲子收集于干燥、洁净、有盖的容器中。大量泄漏:用塑料布、帆布覆盖。然后收集回收或运至废物处理场所处置。
苏州大学有机化学实验-邻、对硝基苯胺的制备(一)
苏州大学化学化工学院课程教案 [实验名称] 邻硝基苯胺和对硝基苯胺的制备(一) [教学目标] 学习氨基的保护、芳环上的硝化和酰胺水解的实验方法;学习薄板的制备。[教学重点] 芳环上的硝化、氨基脱保护。 [教学难点] 芳环硝化反应的条件控制。 [教学方法] 讨论法,演示法,讲述法 [教学过程] [引言] 【实验内容】邻硝基苯胺和对硝基苯胺的制备(一) 【实验目的】学习氨基的保护、芳环上的硝化和酰胺水解的实验方法;学习薄板的 制备。 [提问并讲述] 【实验原理】以乙酰苯胺为原料,通过硝化、水解得到邻硝基苯胺和对硝基苯胺的混合物,以此来验证芳环上的亲电取代反应的定位规律,巩固硝化反应、酰胺的水解 等基本有机反应。反应式如下: NHCOCH3 +HNO3H2SO4 NHCOCH3NHCOCH3 + NO2 2 NHCOCH3NHCOCH3 + NO2 2NH2NH2 + NO2 2 +H2O KOH +CH3COOK [讲述] 苯胺因其极易被氧化,故不能用混酸直接进行硝化,但是可以通过先将氨基保护后硝化,最后脱保护的方法来实现硝基苯胺的合成,本实验以乙酰苯胺为原料来制 备邻、对硝基苯胺。 [讲述] 【实验装置图】
【实验步骤】 一. 乙酰苯胺的硝化 在50 mL三颈瓶中放入2.7 g(0.02 mmol)乙酰苯胺,加入8 mL冰醋酸[1]。安装上电磁搅拌装置。在三颈瓶口分别装上温度计、回流冷凝管、恒压漏斗[2]。在恒压漏斗中加入2.0 mL浓硝酸(比重为1.14 g/mL,0.03 mol)和4 mL浓硫酸(比重为 1.84 g/mL)的混合液[3]。三颈瓶外用水浴控温在50±5 o C[4](理论上如此,实际控温最 好不低于55℃,太低反应难开始而造成积聚,一旦开始又过于剧烈发生冲料;控温在55-60℃较为适宜),边搅拌边慢慢加入混酸(约需20 min,注意恒压滴液漏斗是否积液,控制滴加速度,若反应液变绿则需减缓滴加)[5],加完后在60 o C继续反应1小时(尽量控温不超过70℃,在60-65℃较宜,反应液应为橙红色,淡黄色或绿色都会影响产率)。然后将反应液倒入30 g碎冰中,即有黄色沉淀析出,过滤、冰水洗至近中性[6],再用少量石油醚淋洗,晾干,称重,计算产率。(用纸包裹写好名字放置,下次称量后继续使用) 二.邻、对硝基乙酰苯胺的水解 将制得的邻、对硝基乙酰苯胺1.3 g (0.007 mol) 放入25 mL圆底烧瓶中[7],加入 8.5 mL氢氧化钾醇溶液[8],摇匀,加沸石;装上回流冷凝管(接口处涂真空油脂避免粘 连),在水浴上加热回流半小时,然后从冷凝管口加入3 mL热水。继续在沸水浴中加热20 min,稍冷后倒入20 g冰水中,过滤,用冰水洗至弱碱性或中性[9],晾干[10]得水解产物,称重并计算水解产率。
对硝基苯胺的制备
对硝基苯胺的制备 化学一班常拯波201309020103 1前言 对硝基苯胺,黄色针状结晶,高毒,易升华。微溶于冷水,溶于沸水、乙醇、乙醚、苯和酸溶液。广泛应用于染料工业的人工合成化学物,是多种印染及医药化工品的中间体,也可用于分析试剂。操作时需穿戴防护措施,避免释放至环境。 工业生产对硝基苯胺。可采用乙酰苯胺硝化、水解的方法,也可用对硝基氯苯氨解的方法。 1. 以乙酰苯胺为原料,经硝化、水解而制得。原料消耗定额:乙酰苯胺1210kg/t、硝酸(90%)580kg/t、硫酸3620kg/t、液碱(30%)660kg/t。 2. 以对硝基氯苯为原料,可采用高压釜间歇法生产,也可采用管道反应器连续化生产,收率都在94%左右。原料消耗定额:对硝基氯苯(97%)1170kg/t、氨水(28%)700kg/t 高毒。可引起比苯胺更强的血液中毒。如果同时存在有机溶剂或在饮酒后,这种作用更为强烈。急性中毒表现为开始头痛、颜面潮红、呼吸急促,有时伴有恶心、呕吐,之后肌肉无力、发绀、脉搏频弱及呼吸急促。皮肤接触后会引起湿疹及皮炎。大鼠经口LD50为1410mg/kg 2实验目的 1掌握由苯胺设计合成对硝基苯胺的原理 2、掌握邻硝基苯胺和对硝基苯胺的分离方法 3、学会对有毒药品的操作和处理 3反应中各步化合物的物理性质 化合物名称分子量性状熔点℃沸点℃溶解度水乙醇乙醚 苯胺93.12 无色油状液体-6.3 184 微溶溶溶 乙酸酐 102.09 无色透明液体-73.1 138.6 微溶溶溶
乙酰苯胺135.16 斜方晶体133.4 305 微溶于冷水,溶于热水溶溶 对硝基乙酰苯胺180.16 无色晶体100 215.6 微溶于冷水,易溶于沸水溶溶 邻硝基乙酰苯胺180.16 淡黄色片状94 100 微溶于冷水,易溶于沸水溶溶 对硝基苯胺138.12 淡黄色针状148.5 331.7 微溶于冷水,易溶于沸水溶溶 邻硝基苯胺138.12 橙黄色针状69.7 284.5 微溶于冷水,易溶于沸水溶溶 4实验原理 先以苯胺为原料,经乙酰化合成乙酰苯胺,再经过硝化,水解得到邻硝基苯胺和对硝基苯胺的混合物,再通过蒸馏,柱层析,或水蒸气蒸馏分离即可得到对硝基苯胺 1乙酰苯胺的制备 乙酸和苯胺的反应是可逆的,且反应速率较慢,可采用乙酸过量的方法和利用分馏柱将反应中生成的水蒸除,使平衡向水生成的方向移动而提高乙酰苯胺的产率 2、硝化反应
实验六 对硝基苯胺的制备
实验六 对硝基苯胺的制备(一) 一、实验目的 了解芳香族硝基化合物的制备方法,尤其是由芳胺制备芳香族硝基化合物的方法。 二、实验原理 主反应: +CH 3COOH O 2N NH 2H 2SO 4H 2SO 4H 2O +O 2N NHCOCH 3H 2O ++O 2N NHCOCH 3HONO 2 NHCOCH 3 副反应: +CH 3COOH NHCOCH 3NO 2H 2SO 4H 2SO 4 H 2O +NHCOCH 3H 2O ++NH 2HONO 2 NHCOCH 3 三、实验药品 乙酰苯胺5g (0.037mol ),硝酸(d =1.40)2.2mL (0.032mol ),浓硫酸,冰醋酸,乙醇,碳酸钠,20%氢氧化钠溶液。 四、实验仪器 锥形瓶,烧杯,滴管,抽滤装置。 五、实验步骤 1.对硝基乙酰苯胺的制备 在100ml 锥形瓶内,放入5g 乙酰苯胺和5mL 冰醋酸[1]。用冷水冷 却,一边摇动锥形瓶,一边慢慢地加入 10mL 浓硫酸。乙酰苯胺逐 渐溶解。将所得溶液放在冰盐浴中冷却到0~2℃。 在冰盐浴中用2.2mL 浓硝酸和1.4mL 浓硫酸配置混酸。一边摇动 锥形瓶,一边用吸管慢慢地滴加此混酸,保持反应温度不超过5℃ [2]。 从冰盐浴中取出锥形瓶,在室温下放置30min ,间歇摇荡之。在搅 拌下把反应混合物以细流慢慢地倒入20mL 水和20g 碎冰的混合物 中,对硝基乙酰苯胺立刻成固体析出。放置约10min ,减压过滤, 尽量挤压掉粗产品中的酸液,用冰水洗涤三次,每次用10mL 。称 取粗产品0.2g (样品A ),放在空气中晾干。其余部分[3]用95%乙醇进行重结晶[4]。减压过滤从乙醇中析出的对硝基乙酰苯胺,用少许冷乙醇洗涤,尽量压挤去乙醇。将得到的对硝基乙酰苯胺(样品B )放在空气中晾干。 将所得乙醇母液在水浴上蒸发到其原体积的2/3。如有不溶物,减压过滤。保存母液(样品
对硝基苯甲酸的制备
对硝基苯甲酸的制备(预习报告) 一、实验目的 1、掌握利用对硝基甲苯制备对硝基苯甲酸的原理及方法。 2、掌握电动搅拌装置的安装及使用。 3、练习并掌握固体酸性产品的纯化方法。 二、实验原理 该反应为两相反应,还要不断滴加浓硫酸,为了增加两相的接触面,为了尽可能使其迅速均匀地混合,以避免因局部过浓、过热而导致其它副反应的发生或有机物的分解,本实验采用电动搅拌装置。这样不但可以较好地控制反应温度,同时也能缩短反应时间和提高产率。 生成的粗产品为酸性固体物质,可通过加碱溶解、再酸化的办法来纯化。纯化的产品用蒸汽浴干燥。 三、实验药品用量及物理常数
四、实验装置图 五、实验流程及步骤 1.安装带搅拌、回流、滴液的装置如图 2.在250ml的三颈瓶中依次加入6g对硝基甲苯,18g重铬酸钾粉末及40ml 水。 3.在搅拌下自滴液漏斗滴入25ml浓硫酸。(注意用冷水冷却,以免对硝基甲苯因温度过高挥发而凝结在冷凝管上)。 4.硫酸滴完后,加热回流,反应液呈黑色。(此过程中,冷凝管可能会有白色的对硝基甲苯析出,可适当关小冷凝水,使其熔融滴下)。 5.待反应物冷却后,搅拌下加入80ml冰水,有沉淀析出,抽滤并用50ml 水分两次洗涤。 6.将洗涤后的对硝基苯甲酸的黑色固体放入盛有30ml 5%硫酸中,沸水浴上加热10min,冷却后抽滤。(目的是为了除去未反应完的铬盐)7.将抽滤后的固体溶于50ml 5%NaOH溶液中,50℃温热后抽滤,在滤液中加入1g活性炭,渚沸趁热抽滤。(此步操作很关键,温度过高对硝基甲苯融化被滤入滤液中,温度过低对硝基苯甲酸钠会析出,影响产物的纯度或产率) 8.充分搅拌下将抽滤得到的滤液慢慢加入盛有60ml 15%硫酸溶液的烧杯中析出黄色沉淀,抽滤,少量冷水洗涤两次,干燥后称重。(加入顺序不能颠倒,否则会造成产品不纯)。 9.混合溶剂重结晶粗对硝基苯甲酸。
由苯胺出发合成对硝基苯胺(论文)
由苯胺出发合成对硝基苯胺 姓名:刘昌彬、余婵 大连大学环境与化学工程学院化学 指导老师:李争宁 摘要:利用苯氨经酰基化,硝化,水解制备对硝基苯胺,研究了反应物物料比例,反应温度、反应时间对产物对硝基苯胺的产率的影响。以得到制备对硝基苯胺的产率最高的反应物比例方案。 关键字:苯胺,对硝基苯胺 1.前言: 对硝基苯胺,黄色针状结晶,高毒,易升华。微溶于冷水,溶于沸水、乙醇、乙醚、苯和酸溶液。广泛应用于染料工业的人工合成化学物,是多种印染及医药化工品的中间体,也可用于分析试剂。操作时需穿戴防护措施,避免释放至环境。 2.实验 2、1实验原理 先以苯胺为原料,经乙酰化合成乙酰苯胺,再经过硝化,水解得到邻硝基苯胺和对硝基苯胺的混合物,再通过蒸馏,柱层析,或水蒸气蒸馏分离即可得到对硝基苯胺。 2.2主要试剂及仪器 试剂:苯胺,乙酸酐,冰醋酸,浓硝酸,浓硫酸,NaOH固体,活性炭 仪器:圆底烧瓶,锥形瓶,烧杯250mL,玻璃棒,冷凝管,橡胶管,布氏漏斗,滤纸,铁架台,加热炉 2.3合成方法 3.3.1合成路线 1、乙酰苯胺的制备 乙酸和苯胺的反应是可逆的,且反应速率较慢,可采用乙酸过量的方法和利用分馏柱将反应中生成的水蒸除,使平衡向水生成的方向移动而提高乙酰苯胺的产率。 2、硝化反应 乙酰苯胺与混酸反应,硝化的位置与温度有关。在低温(低于5℃)下产物以对硝基乙酰苯胺为主。硝化温度升高,邻硝基乙酰苯胺产物将增多。 3、水解反应:
pH=10时,邻位产物较对位产物易水解,生成的邻硝基苯胺又溶于50℃的碱液,故将混合产物与碳酸钠溶液共沸水解,50℃过滤即可除去邻位副产物。对位产物再与氢氧化钠溶液共沸,水解得对硝基苯胺。 4、邻硝基苯胺和对硝基苯胺的分离 分馏:利用对硝基苯胺和邻硝基苯胺沸点的差异,采用分馏的方法。 2.3.2合成步骤 1、乙酰苯胺的合成 将5mL苯胺和10mL冰醋酸加入50mL圆底烧瓶中,再取6mL乙酸酐在搅拌下加入圆底烧瓶中,接上直型冷凝管,开通冷凝水,回流15min左右,待反应体系颜色接近橙黄色后,移开热源,从冷凝管口加入5mL蒸馏水,再回流5min。反应结束,在搅拌下趁热将反应物倒入盛有30mL水的烧杯中,待体系冷却后用蒸馏水洗涤抽干。重结晶,向250mL烧杯中加入100mL水和刚得到的产品,加热搅拌至产品全溶。若出现熔化呈油滴现象,就继续加热。预热一个布氏漏斗,趁热抽滤,得到部分产品,再将溶液冷却,最终析出晶体,洗涤,抽滤也是乙酰苯胺产品。 2、乙酰苯胺的硝化 将2.4g已干燥研细的自制乙酰苯胺和4.0mL冰乙酸放入50mL锥形瓶中,充分摇动,然后在冰水浴冷却下慢慢加入5mL浓硫酸,并放在冰浴中冷却至0℃左右。 在另一个 25mL 锥形瓶中配制混酸2.0mL浓硫酸和1.5mL浓硝酸。先加入浓硫酸,然后在冰浴中,边摇动锥形瓶边小心加入硝酸。冷至室温后,用滴管逐滴加到盛放乙酰苯胺溶液的锥形瓶中进行硝化。边加混酸边冷却,使体系温度始终低于 5C。混酸加完后,把锥形瓶移出冰浴,在室温中保持 30min,并间歇摇动锥形瓶。 在搅拌下,将硝化物料细流倒入盛有 25mL 水和25g碎冰的烧杯中,搅拌 5min,抽滤。压干滤饼,用适量冰水洗涤两次后抽干。 将滤饼放入250mL 锥形瓶中,加20~30mL水。在摇动下小心加入碳酸钠粉末,滴加几滴酚酞直至溶液呈微红色。加热至沸腾并保持5min ,再冷却到50℃ ,迅速抽滤。并用适量冰水洗涤两次后抽干。取少量样品重结晶,测定其熔点。
对硝基乙酰苯胺的制备与纯化
对硝基苯胺的制备与纯化 杨丽 2011301040057 化学基地班(三) 摘要:本实验用乙酰苯胺作为起始原料,首先经过硝化获得了邻硝基乙酰苯胺和对硝基乙酰苯胺的混合物。接着采取了两种分离纯化的途径:一种是先将制得的混合物经过重结晶后获得纯的对硝基乙酰苯胺,通过测定固体的熔点来验证所得对硝基乙酰苯胺的纯度。再将重结晶后的产品用氢氧化钠的醇溶液水解,取水解后的产品作薄层析和熔点测定来确定水解后的产物。 另一种方法是直接将硝化后的混合物水解,制得邻硝基苯胺和对硝基苯胺的混合物。通过薄层分析和熔点的测定来证实两者的存在。再分别用重结晶和水蒸气蒸馏的办法分离纯化所获得的产品。并通过薄层析和测熔点的方法来验证产品的组成和纯度。 最后,根据实验结果分析比较两种分离纯化途径的优劣。 绪言:对硝基苯胺常温下是淡黄色针状结晶,易于升华。熔点 148.5℃,沸点 331.7℃,相对密度1.424(20/4℃)。闪点199°,水中溶解度为0.0008g。微溶于冷水,溶于沸水、乙醇、乙醚、苯和酸溶液,有毒,空气中容许浓度为5mg/m3。吸入、口服和皮肤接触有害。所以在实验过程中要尽量避免与之接触。对硝基苯胺是染料工业极为重要的中间体,可直接用于合成:对苯二胺,邻氯对硝基苯胺, 2.6-二氯-4硝基苯胺,5-硝基-2-氯苯酚等,同时还是防老剂,光稳定剂,显影剂等的原料。可作黑色盐 K,供棉麻织物染色、印花之用。并且可作农药和兽药的中间体,在医药工业中可用于生产氯硝胺、卡巴肿、硝基安定、喹啉脲硫酸盐等。还可用于生产对苯二胺;抗氧化剂和防腐剂等。因此对硝基苯胺的合成具有很大的应用价值。工业上生产对硝基苯胺的方法有乙酰苯胺的硝化水解和对硝基氯苯氨解两种方法。 本实验采用的是乙酰苯胺硝化水解的方法,具体步骤将在实验部分给出。反应原理如下:1、乙酰苯胺的硝化 2、硝基乙酰苯胺的水解:
对硝基苯胺的制备
对硝基苯胺的制备(一) 一、 实验目的 了解芳香族硝基化合物的制备方法,尤其是由芳胺制备芳香族硝基化合物的方法。 二、 实验原理 主反应: 副反应: 二、实验药品 乙酰苯胺 5g (0.037mol ),硝酸(d=1.40) 2.2mL (0.032mol ),浓硫酸,冰醋酸,乙醇,碳 酸钠,20%氢氧化钠溶液。 四、 实验仪器 锥形瓶,烧杯,滴管,抽滤装置。 五、 实验步骤 1. 对硝基乙酰苯 胺的制备 M 在100ml 锥形瓶内,放入 5g 乙酰苯胺和5mL 冰醋酸⑴。用冷水冷 却,一边摇动锥形瓶,一边慢慢地加入 10mL 浓硫酸。乙酰苯胺逐 j 渐溶解。将所得溶液放在冰盐浴中冷却到 0?2C 。 在冰盐浴中用 2.2mL 浓硝酸和1.4mL 浓硫酸配置混酸。一边摇动 ; 锥形瓶,一边用吸管慢慢地滴加此混酸,保持反应温度不超过 5C ? * 从冰盐浴中取出锥形瓶,在室温下放置 30min,间歇摇荡之。在搅 W, 拌下把反应混合物以细流慢慢地倒入 20mL 水和20g 碎冰的混合物 / \ 中,对硝基乙酰苯胺立刻成固体析出。放置约 10min,减压过滤, I ) 尽量挤压掉粗产品中的酸液,用冰水洗涤三次,每次用 10mL 。称 取粗产品0.2g (样品A ),放在空气中晾干。其余部分 [3]用95%乙醇进行重结晶 囹。减压过 滤从乙醇中析出的对硝基乙酰苯胺, 用少许冷乙醇洗涤, 尽量压挤去乙醇。将得到的对硝基 乙酰苯胺(样品 B )放在空气中晾干。 H 2SO 4 NHCOCH 3 + HONO 2 ------------ - O 2N __ - H 2SO 4 - NHCOCH 3 + H 2O 2― O 2N NHCOCH 3 + H 2O NH 2 + CH 3COOH H 2SO 4 NHCOCH 3 + H 2O ------------- H 2SO 4 NHCOCH 3 + HONO 2 - NH 2 + CH 3COOH NO 2 NHCOCH 3 + H 2O
由苯胺设计合成对硝基苯胺
实验名称由苯胺设计合成对硝基苯胺 院系化学化工学院 班级化基1101 学号 姓名永超
一、实验目的 1、掌握由苯胺设计合成对硝基苯胺的原理 2、掌握邻硝基苯胺和对硝基苯胺的分离方法 3、学会对有毒药品的操作和处理 二、预备知识 1、反应中各步化合物的物理性质
2、酰化反应的反应活性:酰氯>酸酐>酯>酰胺,故乙酰苯胺可由苯胺与酰氯、酸酐或冰醋酸来制备。在芳胺的反应中,常将氨基通过乙酰化反应保护起来,降低了氨基在亲电取代反应中的活化能力,以防止氨基被氧化。由于乙酰基的空间位阻效应,酰胺基属于邻对位定位基,在苯环上往往选择性地生成邻对位取代产物。 三、实验原理 先以苯胺为原料,经乙酰化合成乙酰苯胺,再经过硝化,水解得到邻硝基苯胺和对硝基苯胺的混合物,再通过蒸馏,柱层析,或水蒸气蒸馏分离即可得到对硝基苯胺。 1、乙酰苯胺的制备
乙酸和苯胺的反应是可逆的,且反应速率较慢,可采用乙酸过量的方法和利用分馏柱将反应中生成的水蒸除,使平衡向水生成的方向移动而提高乙酰苯胺的产率。 2、硝化反应 乙酰苯胺与混酸反应,硝化的位置与温度有关。在低温(低于5℃)下产物以对硝基乙酰苯胺为主。硝化温度升高,邻硝基乙酰苯胺产物将增多。 3、水解反应: pH=10时,邻位产物较对位产物易水解,生成的邻硝基苯胺又溶于50℃的碱液,故将混合产物与碳酸钠溶液共沸水解,50℃过滤即可除去邻位副产物。对位产物再与氢氧化钠溶液共沸,水解得对硝基苯胺。 四、实验试剂和仪器
试剂:苯胺,乙酸酐,冰醋酸,浓硝酸,浓硫酸,20%NaOH,活性炭,40%硫酸 仪器:圆底烧瓶,锥形瓶,烧杯250mL,玻璃棒,冷凝管,橡胶管,布氏漏斗,滤纸,铁架台,加热炉 五、实验步骤 1.乙酰苯胺的合成 将5mL苯胺和10mL冰醋酸加入50mL圆底烧瓶中,再取6mL乙酸酐在搅拌下加入圆底烧瓶中,接上直型冷凝管,开通冷凝水,回流15min左右,待反应体系颜色接近橙黄色后,移开热源,从冷凝管口加入5mL蒸馏水,再回流5min。反应结束,在搅拌下趁热将反应物倒入盛有30mL水的烧杯中,待体系冷却后用蒸馏水洗涤抽干。重结晶,向250mL烧杯中加入100mL水和刚得到的产品,加热搅拌至产品全溶。若出现熔化呈油滴现象,就继续加热。预热一个布氏漏斗,趁热抽滤,得到部分产品,再将溶液冷却,最终析出晶体,洗涤,抽滤也是乙酰苯胺产品。 2、乙酰苯胺的硝化 将2.4g已干燥研细的自制乙酰苯胺和4.0mL冰乙酸放入50mL锥形瓶中,充分摇动,然后在冰水浴冷却下慢慢加入5mL浓硫酸,并放