CRISPR cas9基因敲除原理及其应用

CRISPR/Cas9基因敲除原理及其应用
CRISPR(clustered,regularly interspaced,short palindromic repeats)是一种来自细菌降解入侵的病毒DNA或其他外源DNA的免疫机制。在细菌及古细菌中,CRISPR系统共分成3类,其中Ⅰ类和Ⅲ类需要多种CRISPR相关蛋白(Cas蛋白)共同发挥作用,而Ⅱ类系统只需要一种Cas蛋白即可,这为其能够广泛应用提供了便利条件[1]。
目前,来自Streptococcus pyogenes的CRISPR-Cas9系统应用最为广泛。Cas9蛋白(含有两个核酸酶结构域,可以分别切割DNA两条单链。Cas9首先与crRNA及tracrRNA结合成复合物,然后通过PAM序列结合并侵入DNA,形成RNA-DNA复合结构,进而对目的DNA双链进行切割,使DNA双链断裂。
由于PAM序列结构简单(5’-NGG-3’),几乎可以在所有的基因中找到大量靶点,因此得到广泛的应用。CRISPR-Cas9系统已经成功应用于植物、细菌、酵母、鱼类及哺乳动物细胞,是目前最高效的基因组编辑系统[1]。
通过基因工程手段对crRNA和tracrRNA进行改造,将其连接在一起得到sgRNA(single guide RNA)。融合的RNA具有与野生型RNA类似的活力,但因为结构得到了简化更方便研究者使用。通过将表达sgRNA的原件与表达Cas9的原件相连接,得到可以同时表达两者的质粒,将其转染细胞,便能够对目的基因进行操作[2,3]。
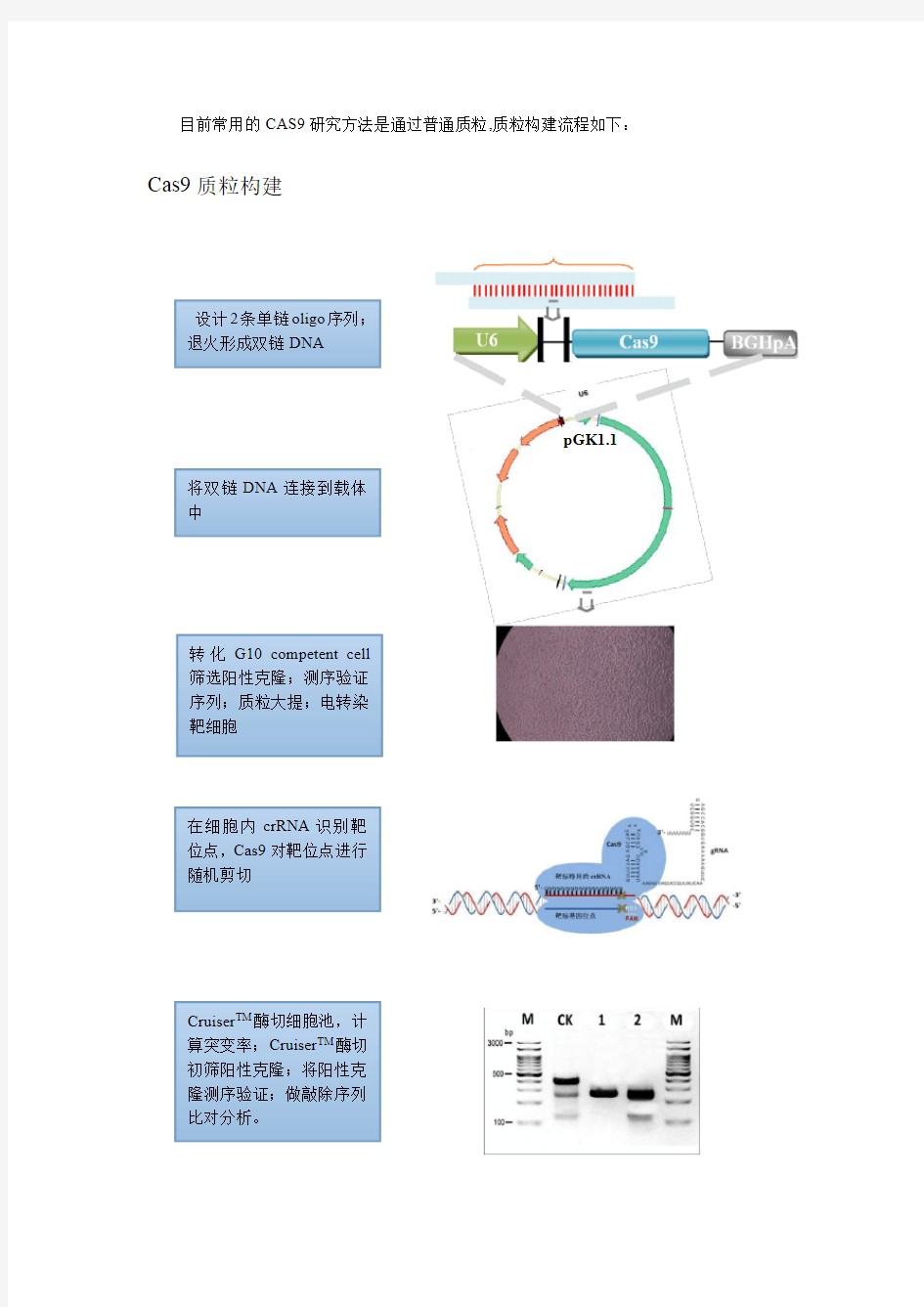
目前常用的CAS9研究方法是通过普通质粒,质粒构建流程如下:Cas9质粒构建
设计2条单链oligo序列;
退火形成双链DNA
pGK1.1
将双链DNA连接到载体
中
转化G10competent cell
筛选阳性克隆;测序验证
序列;质粒大提;电转染
靶细胞
在细胞内crRNA识别靶
位点,Cas9对靶位点进行
随机剪切
Cruiser TM酶切细胞池,计
算突变率;Cruiser TM酶切
初筛阳性克隆;将阳性克
隆测序验证;做敲除序列
比对分析。
目前常见的CAS9普通质粒有(汉恒生物提供cas9质粒试剂盒):
虽然普通质粒很多时候也能达到实验效果,但是质粒转染具有效率低,作用时间短暂性等缺点。病毒的出现解决了质粒这些问题,常用的病毒主要有慢病毒和腺病毒,慢病毒常用质粒见addgene(lentiCRISPR v2,lentiGuide-Puro,lentiCas9-Blast),慢病毒可以整合入宿主基因组中,长期稳定的表达(汉恒生物提供CRISPR/cas9慢病毒包装),但是由于慢病毒克隆能力有限而CAS9本身分子量比较大(大于4kb),且长期插入可能导致乱切,脱靶等,同时慢病毒包装最终获得的滴度不高等原因,腺病毒更有优势,腺病毒克隆能力强,获得的病毒滴度也高。同时相对于普通质粒来说,作用是时间也比较长,可以达到更理想的敲除效果。(汉恒生物提供CRISPR/cas9腺病毒包装)
crispr-cas9基因敲除
CRISPR/Cas9 是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御,可用来对抗入侵的病毒及外源DNA。CRISPR/Cas9 系统通过将入侵噬菌体和质粒DNA 的片段整合到CRISPR 中,并利用相应的CRISPR RNAs(crRNAs)来指导同源序列的降解,从而提供免疫性。 原理 此系统的工作原理是crRNA(CRISPR-derived RNA )通过碱基配对与tracrRNA (trans-activating RNA )结合形成tracrRNA/crRNA 复合物,此复合物引导核酸酶Cas9 蛋白在与crRNA 配对的序列靶位点剪切双链DNA。而通过人工设计这两种RNA,可以改造形成具有引导作用的sgRNA (singleguide RNA ),足以引导Cas9 对DNA 的定点切割。 作为一种RNA 导向的dsDNA 结合蛋白,Cas9 效应物核酸酶是已知的第一个统一因子(unifying factor),能够共定位RNA、DNA 和蛋白,从而拥有巨大的改造潜力。将蛋白与无核酸酶的Cas9(Cas9 nuclease-null)融合,并表达适当的sgRNA ,可靶定任何dsDNA 序列,而sgRNA 的末端可连接到目标DNA,不影响Cas9 的结合。因此,Cas9 能在任何dsDNA 序列处带来任何融合蛋白及RNA,这为生物体的研究和改造带来巨大潜力。 应用 基因敲除动物模型一直以来是在活体动物上开展基因功能研究、寻找合适药物作用靶标的重要工具。但是传统的基因敲除方法需要通过复杂的打靶载体构建、ES细胞筛选、嵌合体小鼠选育等一系列步骤,不仅流程繁琐、对技术的要求很高,而且费用大,耗时较长,成功率受到多方面因素的限制。即使对于技术比较成熟的实验室,利用传统技术构建基因敲除大、小鼠一般也需要一年以上。 2013 年1 月份,美国两个实验室在《Science》杂志发表了基于CRISPR-Cas9 技术在细胞系中进行基因敲除的新方法,该技术与以往的技术不同,是利用靶点特异性的RNA 将Cas9 核酸酶带到基因组上的具体靶点,从而对特定基因位点进行切割导致突变。该技术迅速被运用到基因敲除小鼠和大鼠动物模型的构建之中。通过一系列研究,首先证明了通过RNA 注射的方式将CRISPR-Cas 系统导入小鼠受精卵比DNA 注射能更有效的在胚胎中产生定点突变。在此基础上,又发现了该方法没有小鼠遗传品系的限制,能够对大片段的基因组DNA 进行删除,也可以通过同时注射针对不同基因的RNA 序列达到在同一只小鼠或大鼠中产生多个基因突变的效果。此外,还证明了利用CRISPR-Cas 技术构建的基因敲除大鼠模型与传统方法构建的同一基因(肥胖相关G 蛋白偶联受体Mc4R)突变大鼠相比具有一致的表型。该方法构建的基因突变动物具有显著高于传统方法的生殖系转移能力,是一种可靠、高效、快速的构建敲除动物模型的新方法。 CRISPR-Cas 技术是继锌指核酸酶(ZFN)、ES 细胞打靶和TALEN 等技术后可用于定点构建基因敲除大、小鼠动物的第四种方法,且有效率高、速度快、生殖系转移能力强及简单经济的特点,在动物模型构建的应用前景将非常广阔。 技术优缺点 CRISPR (Clustered Regularly Interspersed Short Palindromic Repeats)是细菌用来抵御病毒侵袭/躲避哺乳动物免疫反应的基因系统。科学家们利用RNA引导Cas9核酸酶可在多种细胞(包括iPS)的特定的基因组位点上进行切割,修饰。Rudolf Jaenisch 研究组将Cas9与Te1和Tet2特异的sgRNA共注射到小鼠的受精卵中,成功得到双基因敲除的纯合子小鼠,效率高达80%。他们将Cas9/sgRNA 与带突变序列的引物共注射,能准确在小鼠两个基因引入所要的点突变。在ES细胞中他们更是成功的一次敲除了五个基因。与ZFN/TALEN相比,CRISPR/Cas更易于操作,效率更高,更容易得到纯合子突变体,而且可以在不同的位点同时引入多个突变。但该系统是否有脱靶效应尚需进一步的研究。 传统的转基因和基因打靶技术,由于技术稳定成熟,可以对小鼠和大鼠的基因组序列进行各种修饰,仍将是模式动物的构建的主要技术。核酸酶ZFN/TALEN 尤其是CRISPR/Cas技术如果能解决脱靶效应的话,有可能会广泛应用于小鼠,大鼠及其他模式动物的制备和研究中,成为传统的转基因和基因打靶技术的重要补充。
CRISPR cas9基因敲除原理及其应用
CRISPR/Cas9基因敲除原理及其应用 CRISPR(clustered,regularly interspaced,short palindromic repeats)是一种来自细菌降解入侵的病毒DNA或其他外源DNA的免疫机制。在细菌及古细菌中,CRISPR系统共分成3类,其中Ⅰ类和Ⅲ类需要多种CRISPR相关蛋白(Cas蛋白)共同发挥作用,而Ⅱ类系统只需要一种Cas蛋白即可,这为其能够广泛应用提供了便利条件[1]。 目前,来自Streptococcus pyogenes的CRISPR-Cas9系统应用最为广泛。Cas9蛋白(含有两个核酸酶结构域,可以分别切割DNA两条单链。Cas9首先与crRNA及tracrRNA结合成复合物,然后通过PAM序列结合并侵入DNA,形成RNA-DNA复合结构,进而对目的DNA双链进行切割,使DNA双链断裂。 由于PAM序列结构简单(5’-NGG-3’),几乎可以在所有的基因中找到大量靶点,因此得到广泛的应用。CRISPR-Cas9系统已经成功应用于植物、细菌、酵母、鱼类及哺乳动物细胞,是目前最高效的基因组编辑系统[1]。 通过基因工程手段对crRNA和tracrRNA进行改造,将其连接在一起得到sgRNA(single guide RNA)。融合的RNA具有与野生型RNA类似的活力,但因为结构得到了简化更方便研究者使用。通过将表达sgRNA的原件与表达Cas9的原件相连接,得到可以同时表达两者的质粒,将其转染细胞,便能够对目的基因进行操作[2,3]。
目前常用的CAS9研究方法是通过普通质粒,质粒构建流程如下:Cas9质粒构建 设计2条单链oligo序列; 退火形成双链DNA pGK1.1 将双链DNA连接到载体 中 转化G10competent cell 筛选阳性克隆;测序验证 序列;质粒大提;电转染 靶细胞 在细胞内crRNA识别靶 位点,Cas9对靶位点进行 随机剪切 Cruiser TM酶切细胞池,计 算突变率;Cruiser TM酶切 初筛阳性克隆;将阳性克 隆测序验证;做敲除序列 比对分析。
基因敲除技术的原理、方法和应用
基因敲除技术的原理、方法和应用 2010-01-24 17:03:43 来源:易生物实验浏览次数:6302 网友评论 0 条 1.基因敲除概述 2.实现基因敲除的多种原理和方法: 2.1.利用基因同源重组进行基因敲除 2.2利用随机插入突变进行基因敲 除。 2.3.RNAi引起的基因敲除。 3.基因敲除技术的应用及前景 4.基因敲除技术的缺陷 关键词:基因敲除 1.基因敲除概述: 基因敲除是自80年代末以来发展起来的一种新型分子生物学技术,是通过一定的途径使机体特定的基因失活或缺失的技术。通常意义上的基因敲除主要是应用DNA同源重组原理,用设计的同源片段替代靶基因片段,从而达到基因敲除的目的。随着基因敲除技术的发展,除了同源重组外,新的原理和技术也逐渐被应用,比较成功的有基因的插入突变和iRNA,它们同样可以达到基因敲除的目的。 2.实现基因敲除的多种原理和方法: 2.1.利用基因同源重组进行基因敲除 基因敲除是80年代后半期应用DNA同源重组原理发展起来的。80年代初,胚胎干细胞(ES细胞)分离和体外培养的成功奠定了基因敲除的技术基础。1985 年,首次证实的哺乳动物细胞中同源重组的存在奠定了基因敲除的理论基础。到1987年,Thompsson首次建立了完整的ES细胞基因敲除的小鼠模型 [1]。直到现在,运用基因同源重组进行基因敲除依然是构建基因敲除动物模型中最普遍的使用方法。 2.1.1利用同源重组构建基因敲除动物模型的基本步骤(图1): a.基因载体的构建:把目的基因和与细胞内靶基因特异片段同源的DNA 分子都重组到带有标记基因(如neo 基因,TK 基因等)的载体上,成为重组载体。基因敲除是为了使某一基因失去其生理功能,所以一般设计为替换型载体。
B-Cas9敲进小鼠
B-Cas9敲进小鼠——一个神奇的基因敲除小鼠制备工具CRISPR/Cas9技术自问世以来就受到了科学家们的青睐。随着基因敲除技术不断更新完善,最近诞生了一种可以快速、高效制备基因敲除小鼠的工具模型--B-Cas9敲进小鼠。B-Cas9敲进小鼠自身携带Cas9蛋白,只需注射sgRNA就可以对基因进行编辑。那么,我们为什么需要B-Cas9敲进小鼠?它有哪些优势、应用领域如何呢?B-Cas9敲进小鼠又是如何制备的呢? 1)B-Cas9敲除小鼠的优势 不仅可以缩短敲除小鼠的制备周期,还可以提高敲除效率。在携带有Cas9蛋白的小鼠体内注射sgRNA病毒,便可即时获得敲除小鼠;若在小鼠体内注射多个sgRNA病毒,就可以实现多基因同时敲除。 2)B-Cas9敲除小鼠的应用 不仅可以快速制备组织特异性基因敲除小鼠模型;其多基因变异更接近于肿瘤的异质性,还可以制备多基因敲除的癌症模型;除此之外, B-Cas9敲进小鼠还可以结合sgRNA文库进行高通量筛选。 3)B-Cas9敲除小鼠的制备原理 将表达Cas9蛋白的序列插入到Rosa26位点当中,并在Cas9前面添加TetO元件,以达到控制Cas9表达,实现可诱导型、组织特异性表达的效果。B-Cas9敲进小鼠只有在注射了Dox后,Dox与Teto复合体结合才能诱导Cas9蛋白的表达。(打靶策略如下图)
4)B-Cas9敲进小鼠与国外引进的Cas9敲进小鼠作比较,我们不难发现,B-Cas9敲进小鼠更加有优势。时间上省去了运输和隔离的3-4个月,费用也减少一半多。从遗传背景和技术改造来看,B-Cas9敲进小鼠的遗传背景是纯净的C57BL/6背景,而国外引进的Cas9敲进小鼠是129背景上回交的。B-Cas9敲进小鼠最大的优势在于其可诱导、可逆性表达Cas9,让实验更加方便,易于操控。
基因敲除技术样本
基因敲除技术 点击次数: 2605 发布日期: -5-25 来源: 本站仅供参考, 谢 绝转载, 否则责任自负 1.概述: 基因敲除是自80年代末以来发展起来的一种新型分子生物学技术, 是经过一定的途径使机体特定的基因失活或缺失的技术。一般意义上的基因敲除主要是应用DNA同源重组原理, 用设计的同源片段替代靶基因片段, 从而达到基因敲除的 目的。随着基因敲除技术的发展, 除了同源重组外, 新的原理和技术也逐渐被应用, 比较成功的有基因的插入突变和iRNA, 它们同样能够达到基因敲除的目的。2.实现基因敲除的多种原理和方法: 2.1.利用基因同源重组进行基因敲除 基因敲除是80年代后半期应用DNA同源重组原理发展起来的。80年代初, 胚胎干细胞( ES细胞) 分离和体外培养的成功奠定了基因敲除的技术基础。1985年, 首次证实的哺乳动物细胞中同源重组的存在奠定了基因敲除的理论基础。到 1987年, Thompsson首次建立了完整的ES细胞基因敲除的小鼠模型[1]。直到现在, 运用基因同源重组进行基因敲除依然是构建基因敲除动物模型中最普遍的 使用方法。 2.1.1利用同源重组构建基因敲除动物模型的基本步骤(图1):
图1.基因同源重组法敲除靶基因的基本步骤 a.基因载体的构建: 把目的基因和与细胞内靶基因特异片段同源的DNA 分子 都重组到带有标记基因(如neo 基因, TK 基因等)的载体上, 成为重组载体。基因敲除是为了使某一基因失去其生理功能, 因此一般设计为替换型载体。 b.ES 细胞的获得: 现在基因敲除一般采用是胚胎干细胞, 最常见的是鼠, 而兔, 猪, 鸡等的胚胎干细胞也有使用。常见的鼠的种系是129及其杂合体, 因为这类小鼠具有自发突变形成畸胎瘤和畸胎肉瘤的倾向, 是基因敲除的理想实验动物。而其它遗传背景的胚胎干细胞系也逐渐被发展应用。[2, 3] c.同源重组: 将重组载体经过一定的方式(电穿孔法或显微注射)导入同源的胚胎干细胞(ES cell)中, 使外源DNA与胚胎干细胞基因组中相应部分发生同源重组, 将重组载体中的DNA序列整合到内源基因组中, 从而得以表示。一般地, 显微注射命中率较高, 但技术难度较大, 电穿孔命中率比显微注射低, 但便于使用。[4,5] d.选择筛选已击中的细胞: 由于基因转移的同源重组自然发生率极低, 动物的重组概率为10-2~10-5, 植物的概率为10-4~10-5。因此如何从众多细胞中筛出真正发生了同源重组的胚胎干细胞非常重要。当前常见的方法是正负筛选法( PNS法) , 标记基因的特异位点表示法以及PCR法。其中应用最多的是PNS法。[6]
基于CRISPR Cas9技术基因敲除小鼠(Cas9-KO)的制作方法-2018-2-28
基于CRISPR Cas9技术基因敲除小鼠(Cas9-KO)的制作方法 一、CRISPR/Cas9靶向基因敲除小鼠制作的基本技术原理: 通过CRISPR/Cas9基因敲除技术,crRNA通过碱基配对与tracrRNA(trans-activating RNA)结合,形成双链RNA。这一tracrRNA:crRNA二元复合体指导Cas9蛋白在crRNA引导序列靶标的特定位点剪切双链DNA。在与crRNA引导序列互补的位点,Cas9蛋白的HNH核酸酶结构域剪切互补链而Cas9 RuvC-like 结构域剪切非互补链,实现敲除目的基因的功能,制备基因敲除小鼠模型。 二、具体步骤如下: 一)模型制作策略制作:利用生物信息学手段(NCBI&IMPC&MGI),分别仔细分析目的基因敲除后小鼠的生存能力及繁育能力,并结合邻近基因的影响,最终选择合适的敲除区域进行敲除方案的设计,出具相应的制作策略。 二)载体的设计和构建:使用麻省理工学院的CRISPR Design工具 (https://www.360docs.net/doc/6c7365937.html,/),依据中靶Score的高低及脱靶Score的高低设计一对长度为20bp的针对靶标DNA的寡聚核苷酸链序列用于制备sgRNA,并在该靶区域设计引物用于后续阳性小鼠的基因鉴定。 1、制备sgRNA的实验方法步骤: 1)线性化pUC57-GDNA-T7载体 中提pUC57-GDNA-T7载体,用BsaI线性化过夜。胶回收保存备用。 2)引物退火及加磷酸 将上下游引物(干粉)稀释,再进行引物退火及加磷酸。 3)连接&阳性菌落筛选
取步骤二中的加磷酸产物与线性化载体pUC57-GDNA-T7进行连接,该连接反应在干式恒温器中进行。对连接产物进行转化,涂板,37°C培养箱过夜培养。再用PCR&测序的方法筛选阳性克隆,再将测序正确的克隆进行甘油菌保种,-80°C保存备用 4)制备转录模板 以构建好的sgRNA载体为模板进行PCR扩增,将PCR产物切胶回收,回收产物离心后倒掉上清留DNA沉淀,再溶解DNA。再吸1 μl测DNA浓度,浓度应介于300-500ng/μl,OD260/280介于1.8-2.0范围内。 5)最后进行sgRNA转录,将得到的sgRNA测浓度,跑电泳,分装保存。 三)Cas9/sgRNA的显微注射:将转录好的Cas9 mRNA,sgRNA混合使用显微操作仪将混合物显微注射到小鼠受精卵的胞浆中,再将受精卵移植到假孕的母鼠子宫中,等待F0代小鼠出生。 四)F0 小鼠的鉴定:在F0代小鼠出生后5-7天时,采用剪脚趾法标记小鼠,并将剪取鼠尾组织用在靶区域设计的引物进行鉴定,选取PCR阳性的样品进行测序。 五)F0代小鼠的可遗传性检测:将PCR以及测序正确的F0代小鼠与野生型C57BL/6小鼠进行交配,产生F1代小鼠,依据F0代小鼠的鉴定方法对F1代小鼠进行鉴定,获得的阳性F1代杂合子小鼠即可稳定遗传。
CRISPRCas基因敲除原理及其应用
C R I S P R C a s基因敲除原 理及其应用 The Standardization Office was revised on the afternoon of December 13, 2020
CRISPR/Cas9基因敲除原理及其应用 CRISPR(clustered,regularlyinterspaced,shortpalindromicrepeats)是一种来自细菌降解 入侵的病毒DNA或其他外源DNA的免疫机制。在细菌及古细菌中,CRISPR 系统共分成3类,其中Ⅰ类和Ⅲ类需要多种CRISPR相关蛋白(Cas蛋白)共同发挥作用,而Ⅱ类系统只需要一种Cas蛋白即可,这为其能够广泛应用提供了便利条件[1]。 目前,来自Streptococcuspyogenes的CRISPR-Cas9系统应用最为广泛。Cas9蛋白(含有两个核酸酶结构域,可以分别切割DNA两条单链。Cas9首先与crRNA及tracrRNA结合成复合物,然后通过PAM序列结合并侵入DNA,形成RNA-DNA复合结构,进而对目的DNA双链进行切割,使DNA双链断裂。 由于PAM序列结构简单(5’-NGG-3’),几乎可以在所有的基因中找到大量靶点,因此得到广泛的应用。CRISPR-Cas9系统已经成功应用于植物、细菌、酵母、鱼类及哺乳动物细胞,是目前最高效的基因组编辑系统[1]。 通过基因工程手段对crRNA和tracrRNA进行改造,将其连接在一起得到sgRNA(singleguideRNA)。融合的RNA具有与野生型RNA类似的活力,但因为结构得到了简化更方便研究者使用。通过将表达sgRNA的原件与表达Cas9的原件相连接,得到可以同时表达两者的质粒,将其转染细胞,便能够对目的基因进行操作[2,3]。 目前常用的CAS9研究方法是通过普通质粒,质粒构建流程如下: Cas9质粒构建 设计2条单链oligo序列;退火形成双链DNA
使用CRISPR-CAS系统构建可遗传的基因敲除小鼠和大鼠
使用CRISPR-CAS系统构建可遗传的基因敲除小鼠和大鼠致编辑:CRISPR-CAS系统已经成为一种细胞和模式生物中有效的基因编辑技术。我们使用CRISPR-CAS系统,通过同时注入两种单导向RNA靶向Uhrf2,同时带入Cas9 mRNA来诱导小鼠的DNA片段缺失。此外,我们通过一种单一的显微注射方法得到了敲除Mc3R和Mc4R两种基因的大鼠。在小鼠和大鼠中均观察到较高的种系转移效率(突变可遗传)。 成簇有序间隔短回文重复关联蛋白系统(CRISPR-CAS系统)是一种在细菌和古细菌中演变的针对病毒和质粒入侵的基于RNA的后天免疫系统。【Bamboo注:该系统由一段Cas基因(双链DNA核酸酶)加一段特异序列组成,Cas作用为结合导向RNA,切割目的基因,导向RNA为CRISPR序列转录而成,有二级结构】根据作用机制不同,CRISPR-CAS系统目前有三种类型。在第二类型(下文称该系统)中,CRISPR序列转录RNA(crRNA)和反式激活RNA(TraceRNA)结合后有能力引导Cas9核酸内切酶到特定的序列,从而导致目标DNA双链缺失。【Bamboo注:机理:摄取了病毒DNA后,CRISPR 序列在转录产物tracrRNA,表达产物CAS蛋白,RNA酶共同作用下产生导向RNA,导向RNA含有病毒DNA序列,可遗传给下一代,再遇到病毒DNA时将其剪切】之前的研究表明在哺乳动物中有多种基因工程使用RNA介导的Cas9核酸酶系统。最近,使用该系统进行高效基因编辑已经在斑马鱼、小鼠和细菌中实现。几个小组也证明通过该系统介导的在细胞和斑马鱼中基因靶向效率与ZFNs(锌指核酸酶)和TALENs(转录活化因子效应核酸酶)【Bamboo注:另外两种常见DNA编辑方式】相似或较高。虽然已经有报道在单个小鼠胚胎中可使用该系统打乱多个基因,但是在动物体中尚未见该系统介导的突变种系转移。此外,长的特异的基因组DNA片段能否被该系统敲除也是未知的。CRISPR-CAS系统基因打靶在其他哺乳动物模型,例如实验室大鼠的效用,还需要确定。在这里,俺们报告俺们使用该系统在小鼠和大鼠中实现了高效的,可遗传的基因敲除。 图1使用该系统产生基因突变鼠。(a)该系统构建:Spacer-核酸引导序列;DR-分隔核酸引导序列的小重复片段;NLS-核酸定位信号序列。(b) F0代大鼠注射导向RNA后突变体的检测:Mc4r靶向前后(-,+)分别使用T7E1酶切Mc4r F0代大鼠尾基因组PCR产物。箭头为突变带,M是Marker.(c) Mc3r和Mc4r基因的序列。红框为核苷酸替换。右侧显示碱基对序列改变。通过DNA测序分析六组克隆扩增的PCR产物序列。各基因型在六组克隆的发生率列在右侧。 为了测试该系统在小鼠中的活动,初始试验选用了细胞中基因组Th位点已经被有效靶向的小鼠。首先,我们向FVB小鼠品系雄性原核(受精卵核物质融合前)中注入不同浓度的线性DNA(针对特定CrDNA和tracrRNA的人工编码Cas9核酸内切酶基因)(图1a),在子鼠中检测Th位点基因突变情况。同ZFNs(锌指核酸酶)和TALENs (转录活化因子效应核酸酶)相似,该系统诱导的双链DNA缺失主要被NHEJ(非同源
基因敲除小鼠技术
转基因、基因敲入/敲除动物技术已经成为现代生命科学基础研究和药物研发领域不可或缺的重要技术,该技术从上世纪七八十年代诞生以来,已有近四十年的历史,经典技术如DNA原核显微注射、胚胎干细胞显微注射技术一直以来经久不衰,并逐渐从基 础研究实验室转向商业模式,成为一项高度标准化的新兴产业 一、技术介绍与研究进展 转基因、基因敲入/敲除动物技术已经成为现代生命科学基础研究和药物研发领域不可或缺的重要技术,该技术从上世纪七八十年代诞生 以来,至今已有近四十年的历史,经典技术如DNA原核显微注射、胚胎干细胞显微注射技术一直以来经久不衰,在小鼠模型构建方面日趋完善,并且如同剪切酶和抗体等 常规分子生物学试剂的制备技术一样,逐渐从基础研究实验室转向商业模式,成为一项高度标准化的新兴产业,催生了数以百计的创新药物和数以千计的优秀文章。尽管 如此,传统技术仍然存在一些难以克服的缺陷,如步骤繁琐、周期漫长、成功率低、费用高昂等,而ZFN和TALEN等新技术的出现,或有可能将这一局面彻底改变。 二、同源重组技术原理 基因敲除鼠技术是上世纪80年代中后期基于DNA同源重组的原理发展起来的,Capecchi和Smithies在1987年根据同源重组(homologous recombination)的原理,首次实现了ES的外源基因的定点整合(targeted integration),这一技术称为"基 因打靶"(gene targeting)或"基因敲除"(gene knockout),利用这种ES的显微注射就可以制作出基因敲出小鼠(KO Mice: knockout mice);由于这一工作,Capecchi 和Smithies于2007年与Evans分享了诺贝尔医学奖。 同源重组(homologous recombination)定义:是指发生在姐妹染色单体(sister chromatin) 之间或同一染色体上含有同源序列的DNA分子之间或分子之内的重新组合。在基因敲除小鼠制作过程中,需要针对目的基因两端特异性片段设计带有相同片段的
用CRISPR Cas9方案构建双基因敲除鼠, 获得双基因敲除纯合子小鼠的交配方案
双基因敲除小鼠繁殖工作: CRISPR/Cas9方案构建双基因敲除鼠,得到F0杂合子之后,如何建系才能获 得双基因敲除纯合子小鼠?这是经常被问到的问题,下面就简单回答一下。 假设我们的目的基因为A和B,通常用CRISPR/Cas9方法得到的基因敲除鼠 为杂合子,双杂合子小鼠基因型为AaBb,大写字母代表野生型(dominant),小写字母代表突变型(recessive)。得到F0杂合子(AaBb)之后,可以用 以下方案之一来获得双基因敲除纯合子小鼠: 方案一: 1.将双杂合子小鼠(AaBb)与野生鼠(AABB)交配,理论上将得到25%的 野生型(AABB),25%基因A单杂合子(AaBB),25%基因B单杂合子 (AABb)及25%双杂合子小鼠(AaBb)。 2.将所得到的双杂合子小鼠(AaBb)互交(inter-cross),理论上6.25% 的后代将会是双基因敲除纯合子小鼠(aabb),见下图。
3.由于双基因敲除实验中一般都需要单基因敲除动物作为对照,所以在 进行上面小鼠breeding的同时可以将基因A单杂合子(AaBB)互交, 在后代中鉴定出基因A纯合子(aaBB),同样将基因B单杂合子(AABb)互交,在后代中鉴定出基因B纯合子(AAbb)。 方案二: 将双杂合子小鼠(AaBb)与单基因纯合子(如aaBB)交配,所生小鼠 中约25%为基因A纯合子而基因B杂合子(aaBb,见下图左)。然后将 aaBb小鼠互交,理论上后代小鼠中25%为双基因敲除纯合子小鼠 (aabb),见下图右。 需要特别注意的几个问题: 1)上面所讲的方法适用于位于不同的染色体两个基因的基因敲除,如 果两个基因位于同一条染色体上,要通过上述方法得到双基因敲除 纯合子小鼠很难; 2)上述方法有赖于基因特异性的Genotyping PCR assays。在开始set up breeding之前必须将两个目的基因特异性的Genotyping PCR assays 准备好; 3)要事先研究一下目的基因敲除后有无胚胎致死性,是否影响其生长 发育等。当然单基因敲除无胚胎致死性并不表示双基因敲除无胚胎 致死性。
基因敲除
基因敲除技术(组图) 一.概述: 基因敲除是自80年代末以来发展起来的一种新型分子生物学技术,是通过一定的途径使机体特定的基因失活或缺失的技术。通常意义上的基因敲除主要是应用DNA 同源重组原理,用设计的同源片段替代靶基因片段,从而达到基因敲除的目的。随着基因敲除技术的发展,除了同源重组外,新的原理和技术也逐渐被应用,比较成功的有基因的插入突变和iRNA ,它们同样可以达到基因敲除的目的。 二.实现基因敲除的多种原理和方法: 1.利用基因同源重组进行基因敲除 基因敲除是80年代后半期应用DNA 同源重组原理发展起来的。80年代初,胚胎干细胞(ES细胞)分离和体外培养的成功奠定了基因敲除的技术基础。1985年,首次证实的哺乳动物细胞中同源重组的存在奠定了基因敲除的理论基础。到1987年,Thompsson首次建立了完整的ES细胞基因敲除的小鼠模型[1]。直到现在,运用基因同源重组进行基因敲除依然是构建基因敲除动物模型中最普遍的使用方法。 (1)利用同源重组构建基因敲除动物模型的基本步骤(图1): ①.基因载体的构建:把目的基因和与细胞内靶基因特异片段同源的DNA 分子都重组到带有标记基因(如neo 基因,TK 基因等)的载体上,成为重组载体。基因敲除是为了使某一基因失去其生理功能,所以一般设计为替换型载体。 ②.ES 细胞的获得:现在基因敲除一般采用是胚胎干细胞,最常用的是鼠,而兔,猪,鸡等的胚胎干细胞也有使用。常用的鼠的种系是129及其杂合体,因为这类小鼠具有自发突变形成畸胎瘤和畸胎肉瘤的倾向,是基因敲除的理想实验动物。而其他遗传背景的胚胎干细胞系也逐渐被发展应用。[2,3] ③.同源重组:将重组载体通过一定的方式(电穿孔法或显微注射)导入同源的胚胎干细胞(ES cell)中,使外源DNA 与胚胎干细胞基因组中相应部分发生同源重组,将重组载体中的DNA 序列整合到内源基因组中,从而得以表达。一般地,显微注射命中率较高,但技术难度较大,电穿孔命中率比显微注射低,但便于使用。[4,5] ④.选择筛选已击中的细胞:由于基因转移的同源重组自然发生率极低,动物的重组概率为10-2 ~10-5 ,植物的概率为10-4 ~10-5 。因此如何从众多细胞中筛出真正发生了同源重组的胚胎干细胞非常重要。目前常用的方法是正负筛选法(PNS法),标记基因的特异位点表达法以及PCR 法。其中应用最多的是PNS法。[6]
基因敲除小鼠制备的流程
基因敲除小鼠的制备流程 基因敲除小鼠已经成为现代生命科学基础研究和药物研发领域不可或缺的实验动物模型,在生命科学、人类医药和健康研究领域中发挥着重要的作用。基于胚胎干细胞的基因打靶技术、EGE技术(基于Crispr cas9技术)是当下比较火热的基因敲除小鼠制备技术。利用这两种技术制备基因敲除小鼠的流程是什么样的? 一、基于胚胎干细胞的基因打靶技术制备基因敲除小鼠的流程: 1.课题设计,订购课题BAC菌; 2.按照课题设计,完成打靶载体设计和构建; 3.将重组载体电转到胚胎干细胞中,用G418筛选转染后的胚胎干细胞, 得到阳性克隆; 4.进一步通过PCR和southern blot杂交技术(基因敲除小鼠检测金标 准)对上一步得到的阳性克隆进行筛选,得到稳定整合外源基因的胚 胎干细胞阳性克隆; 5.将胚胎干细胞阳性克隆注射到小鼠囊胚中,并植入到假孕小鼠的子宫 内; 6.得到嵌合鼠,并获得F1阳性杂合子小鼠。 基于胚胎干细胞的基因打靶技术制备基因敲除小鼠是目前为止唯一一个 可以满足几乎所有基因组修饰要求的打靶技术,但目前只应用在小鼠的基因敲除上,而且其周期长工作量大。 二、利用EGE技术(基于Crispr cas9技术)制备基因敲除小鼠的流程 1.设计构建识别靶序列的sgRNA;
2.设计构建致靶基因切割的EGE系统载体质粒; 3.利用百奥赛图自主开发的UCA试剂盒对sgRNA/Cas9进行活性检测; 4.设计构建打靶载体; 5.体外转录sgRNA/Cas9 mRNA; 6.小鼠受精卵原核注射sgRNA/Cas9 mRNA和打靶载体; 7.获得Fo代小鼠,利用PCR对Fo代小鼠进行基因型鉴定; 8.获得F1代小鼠,利用PCR和southern blot杂交技术(基因敲除小 鼠检测金标准)对F1代小鼠进行基因型鉴定。 虽然EGE技术(基于Crispr cas9技术)制备基因敲除小鼠看似比基于胚胎干细胞的基因打靶技术制备基因敲除小鼠流程繁琐,其实不然,EGE技术(基于Crispr cas9技术)系统构建简单,基因敲除/敲入效率高,速度快,可实现多基因、多物种基因敲除/敲入,最快2个月即可得到F0代阳性鼠,5个月得到F1F1代杂合子小鼠。
大肠杆菌CRISPR-Cas9系统基因敲除简介
1CRISPR-Cas系统的研究进展 CRISPR(clustered regularly interspaced short palindromic repeats),即串联的、间隔的短回文重复序列,最早在1987年研究大肠杆菌的碱性磷酸酶基因时被发现[1]。随后在细菌和古细菌的基因组中也发现大量存在CRISPR,研究证实它能够保护自身抵御外来病毒和质粒的入侵[2],作用机制是依靠crRNA(CRISPR RNA)和tracrRNA(trans-activating crRNA)结合并引导Cas蛋白对外源DNA进行特异性降解[3]。已发现的CRISPR-Cas系统有三种类型:Ⅰ型,Ⅱ型和Ⅲ型,其中以Ⅱ型最为简单,只需一种Cas蛋白,即通过RNA 介导核心蛋白Cas9识别并切割靶序列,引起DNA双链断裂[2]。 受自然界中CRISPR-Cas系统的启发,主要对来自于化脓性链球菌(Streptococcus pyogenes)的Ⅱ型CRISPR-Cas系统进行人为改造和利用,目前已经将其发展成为一种新型的基因编辑技术,实现基因敲除、插入、定点突变和组合编辑[4],并成功应用于大肠杆菌、酿酒酵母、家蚕、果蝇和人类细胞等[5]。和传统的基因编辑技术相比,这一新技术具有成本低、操作简便、效率高的优点[6]。 2 CRISPR-Cas系统的组成与机制 典型的Ⅱ型CRISPR-Cas系统基因座包含tracrRNA基因、Cas蛋白编码基因(cas9、cas1、cas2和csn2)、CRISPR基因座(引导序列、间隔序列和重复序列)这三个部分[6]。 Ⅱ型CRISPR-Cas系统的作用机制可分为三个阶段,第一是高度可变间隔序列的获得(图1),第二是CRISPR-Cas系统基因座的表达,第三是对外源遗传物质的降解[6](图2)。 Cas1、Cas2和Csn2蛋白与新间隔序列的获得相关。与间隔序列同源的外源遗传物质上的原间隔序列(protospacer),其下游存在一段保守序列,被称为PAM(protospacer adjacent motifs)[7]。当外源噬菌体或质粒首次入侵时,宿主菌通过识别PAM,选取合适的原间隔序列,切割并整合到CRISPR基因座中,形成新的间隔序列。 正常情况下CRISPR基因座的表达水平很低[8],但是当外源噬菌体或质粒再次入侵时,CRISPR基因座的表达水平快速上调,首先转录形成前体crRNA(pre-crRNA),随后在tracrRNA的指导下,由Cas9和核酸内切酶RNaseⅢ加工为含有一个间隔序列和部分重复序列的成熟crRNA。 成熟的crRNA与tracrRNA形成crRNA:tracrRNA复合物,并结合Cas9形成核糖核蛋白复合物,crRNA上的间隔序列与外源DNA的原间隔序列互补配对,从而引导具有核酸内切酶活性的Cas9对DNA双链进行特异性切割。 图1 宿主菌获得新间隔序列的机制[6] Fig.1 New spcacer of CRISPR acquisition
基因敲除技术
基因敲除技术 1.概述: 基因敲除是自80年代末以来发展起来的一种新型分子生物学技术,是通过一定的途径使机体特定的基因失活或缺失的技术。通常意义上的基因敲除主要是应用DNA同源重组原理,用设计的同源片段替代靶基因片段,从而达到基因敲除的目的。随着基因敲除技术的发展,除了同源重组外,新的原理和技术也逐渐被应用,比较成功的有基因的插入突变和iRNA,它们同样可以达到基因敲除的目的。 2.实现基因敲除的多种原理和方法: 2.1. 利用基因同源重组进行基因敲除 基因敲除是80年代后半期应用DNA同源重组原理发展起来的。80年代初,胚胎干细胞(ES细胞)分离和体外培养的成功奠定了基因敲除的技术基础。1985年,首次证实的哺乳动物细胞中同源重组的存在奠定了基因敲除的理论基础。到1987年,Thompsson首次建立了完整的ES细胞基因敲除的小鼠模型[1]。直到现在,运用基因同源重组进行基因敲除依然是构建基因敲除动物模型中最普遍的使用方法。 2.1.1利用同源重组构建基因敲除动物模型的基本步骤(图1): a. 基因载体的构建:把目的基因和与细胞内靶基因特异片段同源的DNA 分子都重组到带有标记基因(如neo 基因,TK 基因等)的载体上,成为重组载体。基因敲除是为了使某一基因失去其生理功能,所以一般设计为替换型载体。 b.ES 细胞的获得:现在基因敲除一般采用是胚胎干细胞,最常用的是鼠,而兔,猪,鸡等的胚胎干细胞也有使用。常用的鼠的种系是129及其杂合体,因为这类小鼠具有自发突变形成畸胎瘤和畸胎肉瘤的倾向,是基因敲除的理想实验动物。而其他遗传背景的胚胎干细胞系也逐渐被发展应用。[2,3] c.同源重组:将重组载体通过一定的方式(电穿孔法或显微注射)导入同源的胚胎干细胞(ES cell)中,使外源DNA与胚胎干细胞基因组中相应部分发生同源重
基因敲除技术
基因敲除技术研究进展综述 摘要:基因敲除在20世纪80年代发展起来后已经应用到许多领域, 如建立人类疾病的转基因动物模型(糖尿病转基因小鼠、神经缺损疾病模型等)。这些疾病模型的建立使研究者可以在动物体内进行疾病的研究: 研究发育过程中各个基因的功能, 研究治疗人类遗传性疾病的途径。 关键字:基因敲除;Cre/LoxP系统;基因载体;生物模型 1.概述: 基因敲除又称为基因打靶, 是指从分子水平上将一个基因去除或替代, 然后从整体观察实验动物,推测相应基因功能的实验方法,是功能基因组学研究的重要研究工具。是自80年代末以来发展起来的一种新型分子生物学技术。 通常意义上的基因敲除主要是应用DNA同源重组原理,用设计的同源片段替代靶基因片段,从而达到基因敲除的目的。随着基因敲除技术的发展,除了同源重组外,新的原理和技术也逐渐被应用,比较成功的有基因的插入突变和iRNA,它们同样可以达到基因敲除的目的。 基因敲除已成为当前医学和生物学研究的最热点与最前沿, 并已对生物学和医学的许多研究领域产生深刻的影响, 成为革命性的研究工具, 具有极其重要的理论意义和实践意义。 基于基因敲除技术对医学生物学研究做出的重大贡献,在该领域取得重大进展的三位科学家,70岁的美国人马里奥?卡佩奇(Mario Capecchi)、82岁的美国人奥利弗?史密西斯(Oliver Smithies)和66岁的英国人马丁?埃文斯(Martin Evans)分享了2007年诺贝尔生理学或医学奖。 2.基因敲出技术发展历史 80年代末期的基因敲除技术为第一代技术,属完全性基因敲除,不具备时间和区域特异性。关于第二代区域和组织特异性基因敲除技术的研究始于1993年。Tsien等于1996年在《Cell》首先报道了第一个脑区特异性的基因敲除动物,被誉为条件性基因敲除研究的里程碑。该技术以Cre/LoxP系统为基础,Cre在哪种组织细胞中表达,基因敲除就发生在哪种组织细胞中。 2000年Shimizu等[于《Science》报道了以时间可调性和区域特异性为标志的
Biomics CRISPR Cas9 基因敲除技术
CRISPR/Cas9 基因敲除技术 CRISPR (ClusteredRegularlyInterspacedShortPalindromicRepeats)RNA,是最近几年才发现的原核生物中的调控RNA,用以抵御病毒和质粒入侵。在II型CRISPR系统中,CRISPR RNA(crRNA)与转录激活crRNA(Trans-activating crRNA, tracrRNA)退火形成的复合物能特异识别基因组序列,引导Cas9核酸内切酶在目的片段生成DNA双链断裂(double-strand breaks, DSBs)。CRISPR-Cas系统的高效基因组编辑功能已被应用于多种生物,包括人、小鼠、大鼠、斑马鱼、秀丽隐杆线虫、植物及细菌。多个科研小组的研究都显示,与锌指核酸酶(ZFNs)和转录激活样效应核酸酶(Transcription activator-like effector nucleases, TALEN)相比较,CRISPR-Cas系统介导的基因组靶向实验在真核细胞中具有相似甚至更高的效率。 Biomics专注于RNA基因调控多年,最新推出基因组编辑工具CRISPR/Cas9专家系统,该系统灵活简单、可以对特定基因组位点进行切割置换,特异性高、细胞毒性低。CRISPR/Cas9系统可广泛应用于基因组工程,如基因抑制,基因敲除,基因敲入,基因修复等。CRISPR-Cas9体系的RNA-DNA识别机制为基因组工程研究提供了一项简便而强大的工具。其最重要的优势是Cas9蛋白可在多个不同的gRNA的引导 下同时靶向多个基因组位点,起到多靶点调控的作用。 z CRISPR与RNAi的区别 目前已经广泛应用的RNAi技术的靶标是mRNA,而CRISPR通过RNA识别DNA序列然后再改变DNA序列,是可以遗传的。由于编码mRNA的DNA序列只占总DNA的极少部分,因此靶向DNA序列的CRISPR的靶标要比RNAi广得多,更有可能筛选出针对某DNA序列的特异CRISPR靶标。
基因敲除方法的比较
方法原理优点不足应用 利用同源重组进行基因敲除利用DNA转化技术, 将含有目的基因和靶 基因同源片段的重组 载体导入靶细胞,通 过载体与靶细胞染色 体上同源序列间的重 组,将外源基因整合 入内源基因组内,使 外源基因得以表达。 通过研究靶细胞或者 个体在目的基因插入 前后遗传特性的改 变,达到研究基因功 能的目的。 具有高度特 异性和方向 性,外源片 段也具有可 操作性 1)操作复杂,实验周期长,费用 偏高;(2)在敲除过程中,被破坏 的常常只是靶基因的部分外显 子而并不是整个编码区,残留 的编码序列有可能组合出新的 未知的功能,这将给表型分析 带来麻烦;(3)对于某些必需基 因,敲除后会造成细胞死亡,也 就无法研究这些必需基因的功 能;(4)由于基因功能上的冗余, 敲掉一个基因并不能造成容易 识别的表型;(5)同一个打靶载 体在不同遗传背景下进行基因 敲除,获得的表型差异很大。 噬菌体的 Cre/Loxp系统 和酿酒质粒的 FLP/FRT系统 利用随机插入突变进行基因敲除利用某些能随机插入 基因序列的病毒,细 菌或其他基因载体, 在目标细胞基因组中 进行随机插入突变, 建立一个携带随机插 入突变的细胞库,然 后通过相应的标记进 行筛选获得相应的基 因敲除细胞 效率高、基 因完全失 活、容易分 离鉴定被插 入引起失活 的目的基因 等 1以农杆菌介导的T-DNA插入 突变只适用于那些容易被 T-DNA转化的植物,且常常会引 起的染色体重排现象,使突变 体表型与T-DNA插入无关而难 以进行遗传学分析。 2转座子插入突变要求转座子 本身较短,容易操作,且对任何 拟敲除区域有较高转座效率等 以农杆菌介导 的T-DNA插入 突变 转座子插入突 变 RNA干扰引起的基因敲除是由与生物体内源 靶基因mRNA 同 源的双链RNA 特异性引发的靶m RNA降解,导致目 的基因表达沉默的一 种反向遗传学技术。 1)特异性强, 针对同源基 因共有序列 的RNAi则 只能导致同 源基因失 活,不影响 其他内源性 mRNA的表 达;(2)效率 高;(3)穿透 性强 1缺乏有效的siRNA载体 2无法彻底去除目的基因 3在哺乳动物中的应用还处于 探索阶段,实验方法不够成熟。 1对于一些敲 除后小鼠在胚 胎时就会死亡 的基因,可以 在体外培养的 细胞中利用 RNAi技术研究 它的功能。 2由于RNAi能 高效特异的阻 断基因的表 达,它成为研 究信号传导通
基因敲出技术研究进展 (1)
兰州交通大学化学与生物工程学院综合能力训练Ⅰ——文献综述 题目:基因敲除技术研究进展 作者:蒋成 学号:201207749 指导教师:谢放 完成日期:2014-7-16
基因敲除技术的研究进展 摘要:本文综述了基因敲除技术的研究进展,对其基本步骤、基因敲除技术的种类及近几年来在真核生物与原核生物中的具体应用现状等进行了阐述,并通过列举实例阐明了该技术的作用和未来的发展前景,最后对该技术在应用过程中存在的一些问题做了简单总结。 关键字:基因敲除技术原核中断系统真核中断系统丝状真菌基因敲除1.引言 1.1概念 基因敲除是80年代后半期应用DNA同源重组原理发展起来的。80年代初,胚胎干细胞(ES细胞)分离和体外培养的成功奠定了基因敲除的技术基础[1]。1985年,首次证实的哺乳动物细胞中同源重组的存在奠定了基因敲除的理论基础。到1987年,Thompsson首次建立了完整的ES细胞基因敲除的小鼠模型[2]。直到现在,运用基因同源重组进行基因敲除依然是构建基因敲除动物模型中最普遍的使用方法。 2012年初的敲除决定表面抗原的猪的模型的成功建立更是为异种器官移植排除了一道重要的障碍,展示了基因敲除技术的美好前景。总之, 基因敲除已成为当前医学和生物学研究的最热点与最前沿,并已对生物学和医学的许多研究领域产生深刻的影响,成为革命性的研究工具, 具有极其重要的理论意义和实践意义[1,3]。 基因敲除是借助分子生物学、细胞生物学和动物胚胎学的方法,通过胚胎干细胞[2]这一特殊的中间环节将小鼠的正常功能基因的编码区破坏,揭示基因功能最直接的手段之一, 因此成为后基因组时代的重要研究方法和内容。通常意义上的基因敲除主要是应用DNA同源重组原理[3],用设计的同源片段替代靶基因片段,从而达到基因敲除的目的。 1.2基本步骤 (1)基因载体的构建:目的基因和与细胞内靶基因特异片段同源的DNA分子都重组到带有标记基因,TK基因等)的载体上,成为重组载体。 (2)ES细胞的获得:现在基因敲除一般采用是胚胎干细胞,最常用的是鼠,而兔,猪,鸡等的胚胎干细胞也有使用,而最常用的鼠的种系是129 [4]及其杂合体,因为这类小鼠具有自发突变形成畸胎瘤和畸胎肉瘤的倾向,是基因敲除的理想实验动物。 (3)同源重组:把目的基因和与细胞内靶基因特异片段同源的DNA 分子都重组到带有标记基因(如neo基因,TK基因等)的载体上,使外源DNA与胚胎干细胞基因组中相应部分发生同源重组,将重组载体中的DNA序列整合到内源基因组中,从而得以表达。 (4)选择筛选已击中的细胞:一般地,筛选使用正、负选择法,比如用G418筛选所有能表达neo 基因的细胞,然后淘汰所有HSV -TK正常表达的细胞,剩下的细胞为命中的细胞。将筛选出来的靶细胞导入鼠的囊胚中,再将此囊胚植人母鼠体内,使其发育成嵌合体小鼠[2,6]。 (5)表型研究:通过观察嵌体小鼠的生物学形状的变化进而了解目的基因变化前后对小鼠的生物学形状的改变,达到研究目的基因的目的。