美国药典溶出介质缓冲液的配制
美国药典-溶出方法
711 DISSOLUTION This general chapter is harmonized with the corresponding texts of the European Pharmacopoeia and/or the Japanese Pharmacopoeia. These pharmacopeias have undertaken not to make any unilateral change to this harmonized chapter. Portions of the present general chapter text that are national USP text, and therefore not part of the harmonized text, are marked with symbols () to specify this fact. This test is provided to determine compliance with the dissolution requirements where stated in the individual monograph for dosage forms administered orally. In this general chapter, a dosage unit is defined as 1 tablet or 1 capsule or the amount specified. Of the types of apparatus described herein, use the one specified in the individual monograph. Where the label states that an article is enteric-coated, and where a dissolution or disintegration test that does not specifically state that it is to be applied to delayed-release articles is included in the individual monograph, the procedure and interpretation given for Delayed-Release Dosage Forms is applied unless otherwise specified in the individual monograph. For hard or soft gelatin capsules and gelatin-coated tablets that do not conform to the Dissolution specification, repeat the test as follows. Where water or a medium with a pH of less than 6.8 is specified as the Medium in the individual monograph, the same Medium specified may be used with the addition of purified pepsin that results in an activity of 750,000 Units or less per 1000 mL. For media with a pH of 6.8 or greater, pancreatin can be added to produce not more than 1750 USP Units of protease activity per 1000 mL. USP R EFERENCE S TANDARDS11—USP Chlorpheniramine Maleate Extended-Release Tablets RS. USP Prednisone Tablets RS. APPARATUS Apparatus 1 (Basket Apparatus) The assembly consists of the following: a vessel, which may be covered, made of glass or other inert, transparent material1; a motor; a metallic drive shaft; and a cylindrical basket. The vessel is partially immersed in a suitable water bath of any convenient size or heated by a suitable device such as a heating jacket. The water bath or heating device permits holding the temperature inside the vessel at 37 ± 0.5 during the test and keeping the bath fluid in constant, smooth motion. No part of the assembly, including the environment in which the assembly is placed, contributes significant motion, agitation, or vibration beyond that due to
片剂溶出度相关知识汇总
溶出度知识总结 溶出度(Dissolution rate)也称溶出速率,是指在规定的溶剂和条件下,药物从片剂、胶囊剂、颗粒剂等固体制剂中溶出的速度和程度。测定固体制剂溶出度的过程称为溶出度试验(Dissolution test),它是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法。药物溶出度检查是评价制剂品质和工艺水平的一种有效手段,可以在一定程度上反映主药的晶型、粒度、处方组成、辅料品种和性质、生产工艺等的差异;在产品发生某些变更后(如处方、生产工艺、生产场所变更和生产工艺放大),确认药品质量和疗效的一致性;也是评价制剂活性成分生物利用度和制剂均匀度的一种有效标准,能有效区分同一种药物生物利用度的差异,因此是药品质量控制必检项目之一。 一般认为,难溶性(一般指在水中微溶或不溶)药物,因制剂处方与生产工艺造成临床疗效不稳定的药物以及治疗量与中毒量相接近的药物(包括易溶性药物),其口服固体制剂质量标准中必须设定溶出度检查项。另外固体制剂的处方筛选及生产工艺流程制订过程中,也需对所开发剂型的溶出度做全面考察。一个可行的溶出度试验法应是在不同时间、地点对同一制剂的溶出度测定或不同的操作者之间的测定都必须达到试验结果具有良好的重现性。为了达到以上目的,必须对溶出度测定试验进行全面充分的研究。 生物药剂学(BCS)分类(美国FDA ): 第1类:高溶解度一高渗透性 第2类:低溶解度一高渗透性 第3类:高溶解度一低渗透性 第4类:低溶解度一低渗透性 高溶解度:单个制剂能在250mL,pH值1.0~8.0介质中溶解——相当于中国药典的“微溶” 高渗透性:绝对生物利用度≥90% 上述分类可以作为设定体外溶出度质量标准的依据,也可用于预测能否建立良好的体内-体外相关性的依据。BSC提示,对于高溶解度、高渗透性(1类)药物及某些情况下的高溶解度、低渗透性(3类)药物,其溶出度在0.1NHCL 中15min时为85%即可保证药物的生物利用度不受溶出的限制,即制剂的行为
溶出度检查法美国药典USP-711
<711> DISSOLUTION 溶出度 (USP39-NF34 Page 540) General chapter Dissolution <711> is being harmonized with the corresponding texts of the European Pharmacopoeia and/or the Japanese Pharmacopoeia. These pharmacopeias have undertaken to not make any unilateral change to this harmonized chapter. 通则<711>溶出度与欧盟药典和日本药典中的相应部分相统一。这三部药典承诺不做单方面的修改。 Portions of the present general chapter text that are national USP text, and therefore not part of the harmonized text, are marked with symbols to specify this fact. 本章中的部分文字为本国USP内容,并没有与其他药典统一。此部分以()标注。 This test is provided to determine compliance with the dissolution requirements where stated in the individual monograph for dosage forms administered orally. In this general chapter, a dosage unit is defined as 1 tablet or 1 capsule or the amount specified. Of the types of apparatus designs described herein, use the one specified in the individual monograph. Where the label states that an article is enteric coated and a dissolution or disintegration test does not specifically state that it is to be applied to delayed-release articles and is included in the individual monograph, the procedure and interpretation given for Delayed-Release Dosage Forms are applied, unless otherwise specified in the individual monograph. 本测试用于检测药品口服制剂的溶出度是否符合各论中的规定。本章中,除另有规定外,单位制剂定义为1片或1粒胶囊。对于本章中所述多种仪器,使用各论中规定的种类。除各论中另有规定外,如果检品是肠溶衣片且各论中的溶出度或崩解时限检查项下没有特别指出适用迟释剂的,使用本章中适用于迟释剂的流程和解释。 FOR DOSAGE FORMS CONTAINING OR COATED WITH GELATIN涂有或包含明胶的剂型 If the dosage form containing gelatin does not meet the criteria in the appropriate Acceptance Table (see Interpretation, Immediate-Release Dosage Forms, Extended-Release Dosage Forms, or Delayed-Release Dosage Forms) because of evidence of the presence of cross-linking, the dissolution procedure should be repeated with the addition of enzymes to the medium, as described below, and the dissolution results should be evaluated starting at the first stage of the appropriate Acceptance Table. It is not necessary to continue testing through the last stage (up to 24 units) when criteria are not met during the first stage testing, and evidence of cross-linking is observed. 如果剂型中含有明胶,其不符合验收表中的标准(见判断,速释制剂,延释制剂,缓释制剂),因为存在明胶交联结合作用,它的溶解过程与外加的媒介酶是重复的,见下面的描述,并且溶解结果可以通过适当的验收表的开始的第一阶段标准进行评估。如果溶出结果不满足第一阶段的测试标准,那么就没有必要继续测试到最后阶段,并且也证明了明胶交联结合作用的存在。
溶出度指导原则
附件1 普通口服固体制剂溶出度试验 技术指导原则 一、前言 本指导原则适用于普通口服固体制剂,包括以下内容:(1)溶出度试验的一般要求;(2)根据生物药剂学特性建立溶出度标准的方法;(3)溶出曲线比较的统计学方法;(4)体内生物等效性试验豁免(即采用体外溶出度试验代替体内生物等效性试验)的一般考虑。 本指导原则还针对药品的处方工艺在批准后发生变更时,如何通过溶出度试验确认药品质量和疗效的一致性提出了建议。附录对溶出度试验的方法学、仪器和操作条件进行了概述。 二、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透。由于药物的溶出和溶解对吸收具有重要影响,因此,体外溶出度试验有可能预测其体内行为。基于上述考虑,建立普通口服固体制剂(如片剂和胶囊)体外溶出度试验方法,有下列作用: 1.评价药品批间质量的一致性; 2.指导新制剂的研发;
3.在药品发生某些变更后(如处方、生产工艺、生产场所变更和生产工艺放大),确认药品质量和疗效的一致性。 在药品批准过程中确定溶出度标准时,应考虑到药物的溶解性、渗透性、溶出行为及药代动力学特性等因素,以保证药品批间质量的一致性、变更以及工艺放大前后药品质量的一致性。 对于新药申请,应提供关键临床试验和/或生物利用度试验用样品以及其他人体试验用样品的体外溶出度数据。对于仿制药申请,应在溶出曲线研究的基础上制定溶出度标准。无论是新药还是仿制药申请,均应根据可接受的临床试验用样品、生物利用度和/或生物等效性试验用样品的溶出度结果,制定溶出度标准。 三、生物药剂学分类系统 根据药物的溶解性和渗透性,推荐以下生物药剂学分类系统(BCS)(Amidon 1995): 1类:高溶解性–高渗透性药物 2类:低溶解性–高渗透性药物 3类:高溶解性–低渗透性药物 4类:低溶解性–低渗透性药物 上述分类原则可作为制定体外溶出度质量标准的依据,也可用于预测能否建立良好的体内-体外相关性(IVIVC)。在37±1℃下,测定最高剂量单位的药物在250mL pH值介于1.0和8.0之间的溶出介质中的浓度,当药物的最高剂量除以以上介质中的药物浓度小于或等于250mL时,可认为是高溶解性药物。一般情
溶出曲线与溶出介质
溶出曲线与溶出介质 研发过程中遇到的问题解答 问题1.请问我们做的一个复方制剂是一个硬胶囊里包含有一个片子和一个软胶囊但是FDA各自推荐了溶出方法片子用的磷缓软胶囊用的盐酸转速和介质体积取样时间点均不一致但是要整粒硬胶囊投药测A-片溶出就按照片A溶出条件取样只检测A溶出曲线这样B软胶囊等于只是考察到了对A的影响但是不会有B的任何数据等于浪费掉了这样原研就要买很多了呀 而且A片子用的900ml磷缓浓度也是不常规的0.07M的而B软胶囊用的推荐介质是盐酸0.01M的那我做A片子剩余的三个介质溶出曲线是按照指导原则0.1N盐酸还是与-B软胶囊盐酸浓度保持一致也用0.01M盐酸呢反之B软胶囊其它介质比如磷缓是用指导原则0.05M的磷缓还是用A所用浓度0.07M呢如果A 是难溶药物介质中还加入了0.5%SDS又该怎么做?由于原研知己非常昂贵因此谨慎起见想咨询下专家的意见 答:很好很现实的问题。如果可能,你可以开发一个新的溶出方法同时适用于两个APIs。当然如果A是低溶解度药,B是高溶解度药,一个介质适用两个APIs有一定难度。如果你是申请美国审批,不管你最终用什么方法,你都必须要提交FDA推荐的方法(n=12),这是硬规定。 对于不同pH介质,如果你证明0.1 N HCl 和0.01 N HCl 的pH 差别对于药物(两个API)的溶解度和药剂的溶出度都影响很小,你可以使用0.01N HCl。FDA不般不会计较,欧洲有可能计较,CFDA我不太理解。如果0.07 M磷酸介质pH 6.8是推荐介质, 你不需要再重复0.05 M pH 6.8. 对于pH 4.5, 你仍可以适用0.05M 的离子强度。
中、美、英三国新版药典溶出度、释放度检查方法比较
中、美、英三国新版药典溶出度、释放度检查方法比较 许鸣镝胡琴 摘要:目的通过对中国药典、美国药典和英国药典中溶出度、释放度测定方法的比较,使广大药物分析工作者了解其异同,为新药开发及进出口检验服务。方法就其历史沿革,最新版所采用的仪器装置和结果判定等方面进行比较和讨论。结果三国药典收载的仪器装置各有异同,结果判定差异较大,应引起注意。结论如何能准确有效地监控药物释放过程,仍是有待深入研究和完善的课题。 关键词溶出度;释放度;中国药典;美国药典;英国药典 中图分类号:R921 文献标识码:E 文章编号:1001-2494(2000)07-0491-04 溶出度和释放度是指药物从片剂、胶囊剂和其它缓释、控释制剂中,在规定介质内,在一定条件下的溶出速度和溶出程度,是评价药物制剂质量的内在指标,是制剂质量控制的重要手段。 在药检工作中,经常要查阅中国药典、美国药典和英国药典。中国药典是我国药品的最高法典,而美国药典和英国药典由于历史悠久,技术先进又具有代表性,在世界各国有较大影响。有些国家没有药典,而以美国药典和英国药典为标准,在世界药品贸易中也常以其标准来要求。综观新版三国药典所收载的溶出度、释放度检查方法在很多方面趋向一致,但又在某些方面存在差别。下面仅就其历史沿革,最新版所采用的仪器装置和结果判定要求等方面进行比较和讨论。 1 历史沿革 1.1 中国药典 中国药典在1985年版引入溶出度检查法时,设定篮法、桨法及类似于流室法的装置等3种装置,1990年版仅保留了前2种装置,1995年版中增订了小杯法装置,并引入了释放度检查法,至2000年版又增加了测定透皮贴剂释放度所需的桨碟装置,方法发展很快。 1.2 美国药典 美国药典自1970年版(第18版)率先引入溶出度检查法,最初只设转篮法装置,且无图例。1975年版(第19版)增加了转篮法的图例,但与现在试验的篮法装置也不相同。1980年版(第20版)增设了桨法装置和改造后的崩解仪两种装置,未给出图例,也无统一的仪器配件尺寸规格。1985年版(第21版)起,规定了与现在溶出度试验所用篮法、桨法相同的装置,并引入释放度检查法,对缓释、肠溶制剂的溶出进行监控,装置与溶出度检查装置相同。1990年版(第22版)在上述规定的基础上,又增加了测定透皮贴剂的三种装置:桨碟法(paddle over disk),筒法(cylinder)及往复碟法(reciprocating disk)。至美国药典1995年版(第23版),用于溶出度、释放度测定的仪器增至7种,而最新版2000年版(第24版)在此基础上又进一步在设备上增加和完善,以适应更多剂型的要求。目前美国药典第24版已成为收载溶出度、释放度测定方法最多,规定最为详尽的药典。 1.3 英国药典 英国药典在1973年版中规定了地高辛片的溶出度和释放度检查,在1988年版引入溶出度检查法,设篮法、桨法两种装置,1993年版增加流室法装置,但未规定药物释放度检查法。在1998年版中,有关内容变化较大,按国际协调会(ICH)提出的要求,将试验片(个)数由5改为6,还增加了透皮贴剂的溶出度测定法(dissolution test for transdermal patches),并相应规定了3种装置。 由三国药典溶出度和释放度检查法的历史沿革可见,随着药品品种的增加,各国药典对药品的溶出度,释放度检查越来越重视,要求也越来越严格。最新版三国药典各论中收载的溶出度、释放度检查品种数量见表1。 表1
usp美国药典结构梳理
USP35-NF-30结构整理 vivi2010-10-02 USP总目录: 1 New Official Text修订文件 加快修订过程包括勘误表,临时修订声明(IRAS),修订公告。勘误表,临时修订声明,修订公告在USP网站上New Official Text部分刊出,勘误表,临时修订公告也会在PF上刊出2front matter前言 药典与处方集增补删减情况,审核人员,辅料收录情况 3凡例
药典, 1标题和修订 2 药典地位和法律认可 3标准复合性 4专论和通则 5 专论组成 6 检验规范和检验方法 7 测试结果 8 术语和定义 9 处方和配药 10 包装存储与标签 4通则 4.1章节列表 4.2一般检查和含量测定(章节编号小于1000)
检查和含量分析的一般要求 检查和含量分析的仪器, 微生物检查,生物检查和含量测定, 化学检查和含量测定, 物理检查和测定 4.3一般信息(章节号大于1000) 5食物补充剂通则 6试剂(试剂,指示剂,溶液等) 7参考表 性状描述和溶解性查询表(按字母顺序) 8食品补充剂各论(字母顺序) 9NF各论(辅料标准) 10 USP各论 11术语 附件:通则的章节中文目录(使用起来比较方便,直接找对应章节号即可)一、通用试验和检定 (1)试验和检定的总要求 1 注射剂 11 参比标准物 (2)试验和检定的装置 16 自动分析方法 21 测温仪 31 容量装置,如容量瓶、移液管、滴定管,各种规格的误差限度
41 砝码和天平 (3)微生物学试验 51 抗菌效力试验 55 生物指示剂:耐受性能试验 61 微生物限度试验 61 非灭菌制品的微生物检查:计数试验 62 非灭菌制品的特定菌检查,如大肠杆菌、金葡菌、沙门氏菌等 71 无菌试验 (4)生物学试验和检定 81 抗生素微生物检定 85 细菌内毒素试验 87 体外生物反应性试验:检查合成橡胶、塑料、高聚物对哺乳类细胞培养的影响 88 体内生物反应性试验:检查上述物质对小鼠、兔iv、ip或肌内植入的影响 91 泛酸钙检定 111 生物检定法的设计和分析 115 右泛醇检定 121 胰岛素检定 141 蛋白质——生物适应试验,用缺蛋白饲料大鼠,观察水解蛋白注射液和氨基酸混合物的作用 151 热原检查法 161 输血、输液器及类似医疗装置的内毒素、热原、无菌检查 171 维生素B12 活性检定 (5)化学试验和检定 A 鉴别试验 181 有机含氮碱的鉴别 191 一般鉴别试验 193 四环素类鉴别 197 分光光度法鉴别试验 201 薄层色谱鉴别试验 B 限量试验
No.4 —— 溶出介质的选用与配制
上海市药品检验所
谢沐风
撰写
【No.4 ——溶出介质的选用与配制】
—— 上海市药品检验所 谢沐风 撰写
1. 溶出介质的选用 建议采用多条溶出曲线对产品的内在品质进行评价。选取原则建议如下: 【普通制剂】 (1)酸性药物制剂 pH 值分别为 1.0 或 1.2、5.5~6.5、6.8~7.5 和水; pH 值分别为 1.0 或 1.2、3.0~5.0、6.8 和水;
(2)中性或碱性药物/包衣制剂 (3)难溶性药物制剂 (4)肠溶制剂 【调释制剂】
pH 值分别为 1.0 或 1.2、4.0~4.5、6.8 和水;
pH 值分别为 1.0 或 1.2、6.0、6.8 和水;
pH 值分别为 1.0 或 1.2、3.0~5.0、6.8~7.5 和水。
2. 其他事项 (1) 以上含有 pH 值范围的,可分别按 0.5 或 1.0 间隔测试,如差异较大,应分别予以关注; 如无明显差异,酌情选择即可。pH 值 1.0 或 1.2,系因各国要求不同。
(2) 在了解了该药物 pKa 值之后,如 pKa±1.0 值未能涵盖于以上各 pH 值中,建议测定 pKa±1.0 值溶出曲线,以更好地把握该药物的溶出特性。
(3) 如能测定更多 pH 值曲线,自然可得到更多关于该制剂内在品质信息;当然工作量亦会 增加。
(4) 无论何种制剂都不建议采用 pH8.0 以上的介质进行表达。如确有必要,应提供充足的理 由。
(5) 某些品种还可根据临床使用情况, 考虑在“含有胃蛋白酶的模拟胃液”和“含有胰酶的模拟 肠液”中的体外溶出情况。 含有胃蛋白酶的模拟胃液,英文全称为 Simulated Gastric Fluid,简写为 SGF。有时 亦可附有下标“sp” ,缩写为 SGF[sp]。其配制方法—— 【中国药典】 取稀盐酸 16.4ml(相当于盐酸 3.84ml) ,加水约 800ml 与胃蛋白酶 10g,
上海市药品检验所 谢沐风 撰写
1
美国药典溶出度4法联合高效液相色谱分析低释放眼用植入制剂
美国药典溶出度4法联合高效液相色谱分析低释放眼用植入制剂 David C. Browne and Shawn Kieselmann* Intertek, 291路东22号,塞勒姆工业园,白宫,美国新泽西州08888 目的 建立美国药典溶出度4法联合高效液相色谱分析低剂量缓释眼用植入制剂的方法 背景 眼部植入制剂是一种体积小,以聚合物为基底的圆盘或者圆柱形的药用制剂,可改善药物进入眼睛后的治疗释放(1)。植入剂一些常见的类型包括巩膜外层植入剂,放置在眼睛的中纬线上;和玻璃体内植入剂,经外科手术放置在眼睛(2)玻璃体内。图1展示了眼睛的解剖图和各种植入剂的放置位置。眼植入物用于治疗各种疾病,如糖尿病视网膜病变和黄斑变性(1)。选择USP4法来测试该类产品的溶出度是由于该法采用层流的方式与体内条件具有良好的相关性,并且开环的配置方式提供的漏槽条件有利于难溶药物的溶出。 实现目标 该方法的建立主要有三个目标。第一,由于眼部植入制剂药物释放浓度低,需要优化色谱条件以获得定量限≤1ng/ml。其次,确定美国药典溶出度4法在测试过程中产生的干扰或吸附。最后,用美国药典溶出度4法联合高效液相色谱法获取溶出曲线。 方法学 实验初期,选取Sotax CE 7 smart溶出仪(开环),pH 7.4的磷酸盐缓冲液作为溶出介质进行实验。泵流速设置为最低流速(1.5ml/min),用以模拟体内条件,因为人体眼部玻璃体的流速大约为2μl/min(3)。一个剂量单位的药物置于铺有玻璃珠的22.6mm的流通池内。溶出液直接在线注入高效液相色谱仪进行分析,几天内每小时采样一次。不推荐使用收集器和自动进样器,因为待分析物与一些塑料会发生化学反应。
欧洲及美国日本药典不同pH溶出介质(缓冲盐溶液)配制
1.1 溶出介质的配制 2.2.1 欧洲药典配制法 (1) pH值1.0~2.2 盐酸溶液 pH值1.0:精密量取盐酸溶液9.0ml,加水稀释至1000ml,摇匀,即得。 其他pH值溶液:量取一定体积的0.2mol/L盐酸液(量取盐酸18.0ml,加水稀释至 1000ml,摇匀,即得),加水稀释至200ml,摇匀,即得。 pH值 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 2.0 2.1 2.2 0.2mol/L盐酸液(ml) 85.0 67.2 53.2 41.4 32.4 26.0 20.4 16.2 13.0 10.2 7.8 (2) pH值3.8~5.8 醋酸盐缓冲液 2mol/L醋酸溶液:取120.0g冰醋酸(经查,冰醋酸密度为1.049g/ml,故体积约为 114ml),加水稀释至1000ml,摇匀,即得。 取下表中规定物质各量,加水溶解并稀释至1000ml,摇匀,即得。 pH值 3.8 4.1 4.3 4.5 4.7 4.9 5.1 5.2 5.3 5.4 5.5 5.8 三水醋酸钠(g) 0.67 1.5 1.99 2.99 3.59 4.34 5.08 5.23 5.61 5.76 5.98 6.23 2mol/L醋酸液(ml) 22.6 19.5 17.7 14.0 11.8 9.1 6.3 5.8 4.4 3.8 3.0 2.1 (3) pH值4.5~8.0 磷酸盐缓冲液 取6.80g磷酸二氢钾,加一定体积0.2mol/L氢氧化钠液(取8.0g氢氧化钠,加水溶解 并稀释至1000ml,即得)和适量水溶解后,再加水稀释至1000ml,摇匀,即得。 pH值 4.5 5.5 5.8 6.0 6.2 6.4 6.6 0.2mol/L氢氧化钠液(ml)0 9.0 18.0 28.0 40.5 58.0 82.0 pH值 6.8 7.0 7.2 7.4 7.6 7.8 8.0 0.2mol/L氢氧化钠液(ml)112.0 145.5 173.5 195.5 212.0 222.5 230.5 2.2.2 美国药典配制法 (1) pH值1.0~2.2 盐酸/氯化钾溶液 同欧洲药典配制法,但其中要溶解入0.75g氯化钾。 (2) pH值2.2~4.0 盐酸/苯二甲酸氢钾溶液 称取2.04g苯二甲酸氢钾,加入一定体积0.2mol/L盐酸液和适量水溶解后,再加水稀 释至200ml,摇匀,即得。 pH值 2.2 2.4 2.6 2.8 3.0 3.2 3.4 3.6 3.8 4.0 0.2mol/L盐酸液(ml) 49.5 42.2 35.4 28.9 22.3 15.7 10.4 6.3 2.9 0.1 (3) pH值4.2~5.8 氢氧化钠/苯二甲酸氢钾溶液 称取2.04g苯二甲酸氢钾,加入一定体积的0.2mol/L氢氧化钠溶液和适量水溶解后, 再加水稀释至200ml,摇匀,即得。 pH值 4.2 4.4 4.6 4.8 5.0 5.2 5.4 5.6 5.8
中国药典溶出方法补充验证内容
溶出度测定 1试验材料 方法验证用样品信息 制剂规格批号批量试制日期试制地点 奥硝唑片250mg B161103 四川科伦药业股份有限 公司(安岳分公司) 表2方法验证用对照品信息 对照名称批号来源纯度 奥硝唑对照品100608-201102中国食品药品检定研究院100% 2溶出方法 溶出介质配制 溶出介质配制方法 pH1.0 量取盐酸9.0mL,加脱气水适量使成1000 mL,溶解,混匀。 溶出测定方法(UV法) 项目pH1.0 方法桨法 转速50rpm/min 溶出体积1000mL 检测波长277nm 3 重复性 供试品溶液:取本品6片,照溶出度测定法(中国药典2015年版四部通则0931第二法),以pH1.0盐酸溶液1000mL为溶出介质,转速为每分钟50转,依法操作,于30分钟时取分别取溶液10ml,弃去5ml滤液,精密量取续滤液1.0mL,置20ml棕色量瓶中,加溶出介质稀释至刻度,混匀,即得。 对照品溶液:精密量取奥硝唑对照品12.5mg,置100ml棕色量瓶中,加溶出介质溶解并稀释至刻度,再精密量取5ml置50ml棕色量瓶中,加溶出介质定容,即得。 取对照品溶液和供试品溶液在277nm处测定吸光度,计算供试品溶液溶出量及溶出量间的RSD值。 4 中间精密度 由不同操作人员采用不同仪器,按照重复性操作过程制备对照品溶液与供试品溶液,计算供试品溶出量。统计重复性与中间精密度共12份样品的溶出量,并计算RSD值,结果见下表。
5方法耐用性 在原溶出条件下,分别微调各种参数,考察溶出度试验结果,结果见下表:A:温度:37.5℃、36.5℃; B:介质体积:990mL、1010mL; C:转速:48转/分钟、52转/分钟。
USP-1092-溶出度试验的开发和验证(中英文对照版)
(1092)溶出度试验的开发和验证【中英文对照版】 INTRODUCTION 前言 Purpose 目的 The Dissolution Procedure: Developmentand Validation <1092> provides a comprehensive approach covering items to considerfor developing and validating dissolution procedures and the accompanyinganalytical procedures. It addresses the use of automation throughout the testand provides guidance and criteria for validation. It also addresses thetreatment of the data generated and the interpretation of acceptance criteriafor immediate- and modified-release solid oral dosage forms. 溶出实验:开发和验证(1092)指导原则提供了在溶出度方法开发和验证过程中以及采用相应分析方法时需要考虑的因素。本指导原则贯穿溶出度实验的全部过程,并对方法提供了指导和验证标准。同时它还涉及对普通制剂和缓释制剂所生成的数据和接受标准进行说明。 Scope 范围 Chapter <1092> addresses the development andvalidation of dissolution procedures, with a focus on solid oral dosage forms.Many of the concepts presented, however, may be applicable to other dosageforms and routes of administration. General recommendations are given with theunderstanding that modifications of the apparatus and procedures as given in USP general chapters need to be justified. <1092>章节讨论了溶出度实验的开发和验证,重点是口服固体制剂。所提出的许多概念也可能适用于其他剂型和给药途径。关于设备和方法的修改部分在USP通则中给出了合理的说明。 The organization of <1092> follows the sequence of actions often performed inthe development and validation of a dissolution test. The sections appear inthe following sequence. 在进行溶解度实验的开发和验证时,常遵循指导原则<1092>,具体内容如下:1. PRELIMINARY ASSESSMENT (FOR EARLY STAGES OF PRODUCTDEVELOPMENT/DISSOLUTION METHOD DEVELOPMENT) 1.前期评估(对产品开发以及溶出度方法开发的前期研究评估) 1.1 Performing Filter Compatibility 1.1滤膜相容性研究 1.2 Determining Solubility and Stability of DrugSubstance in Various Media 1.2原料药在不同溶出介质中溶解度测定和稳定性研究
溶出度对比研究四种溶出介质的比较样本
001【求助】溶出度对比研究-四种溶出介质的比较 作者: wangjianglin332(站内联系TA)发布: -10-21 Sample Text 溶出度作为工艺筛选和质量研究的重要评价指标, 按当前的审评要求必须做详尽的研究, 现今一上市品种的仿制, 溶出度研究碰到比较棘手的问题, 望园中的前辈不吝赐教。 1.主药易溶于水, 溶解度为0.1g/ml, 一剂量( 240mg) 的主药在250ml四种溶出介质( 酸(0.1mol/L盐酸溶液)、 pH=4.5(醋酸-醋酸钠缓冲液)、 pH=6.8(磷酸盐缓冲液)、水) 能完全溶解。胶囊剂崩解时限为3分钟, 胶囊崩解药物即可溶出, 实验结果也表明在水中, 10分钟药物的溶出量即可达到90%以上, 此种情况还是否需要经过溶出曲线的相似性来判定自制样品和市售品溶出行为的一致性? 园中有帖子谈到用ICH指导原则的三步法进行考察, 那么最后一步证明崩解和溶出具有相关性如何考察? 2.溶出度的比较研究一般需考察自制品和市售品在四种溶出介质( 酸(0.1mol/L 盐酸溶液)、 pH=4.5(醋酸-醋酸钠缓冲液)、 pH=6.8(磷酸盐缓冲液)、水) 中的溶出行为, 溶出量的测定方法也一般采用UV法进行测定。本品取样后, 需进行衍生化反应后才有紫外吸收(一般情况紫外最大吸收波长为195nm, 衍生化后为525nm), 但在实验研究过程中我们发现, 介质酸中样品1小时后吸光值在0. 1至0.2的范围, 不符合紫外吸收值的误差范围; 介质pH=4.5(醋酸-醋酸钠缓冲液)中1小时后样品吸光值为0, 推断无衍生化反应发生; 介质pH=6.8(磷酸盐缓冲液)中1小时后样品吸光值也在0.1至0.2的范围内, 不符合紫外吸收值的误差范围。综合推断, 除水以外, 另外三种介质对衍生化反应均有不同程度影响, 溶出量无法准确测定, 此种情况下能否以水一种介质进行溶出度的对比研究? 举报删除此信息caoyuan521(站内联系TA) 问题一: 在国内一般有溶出检测项目的制剂都不再检测崩解项目了。我个人认为在质量标注中指定了溶出的指标就不要研究与崩解的关系了。
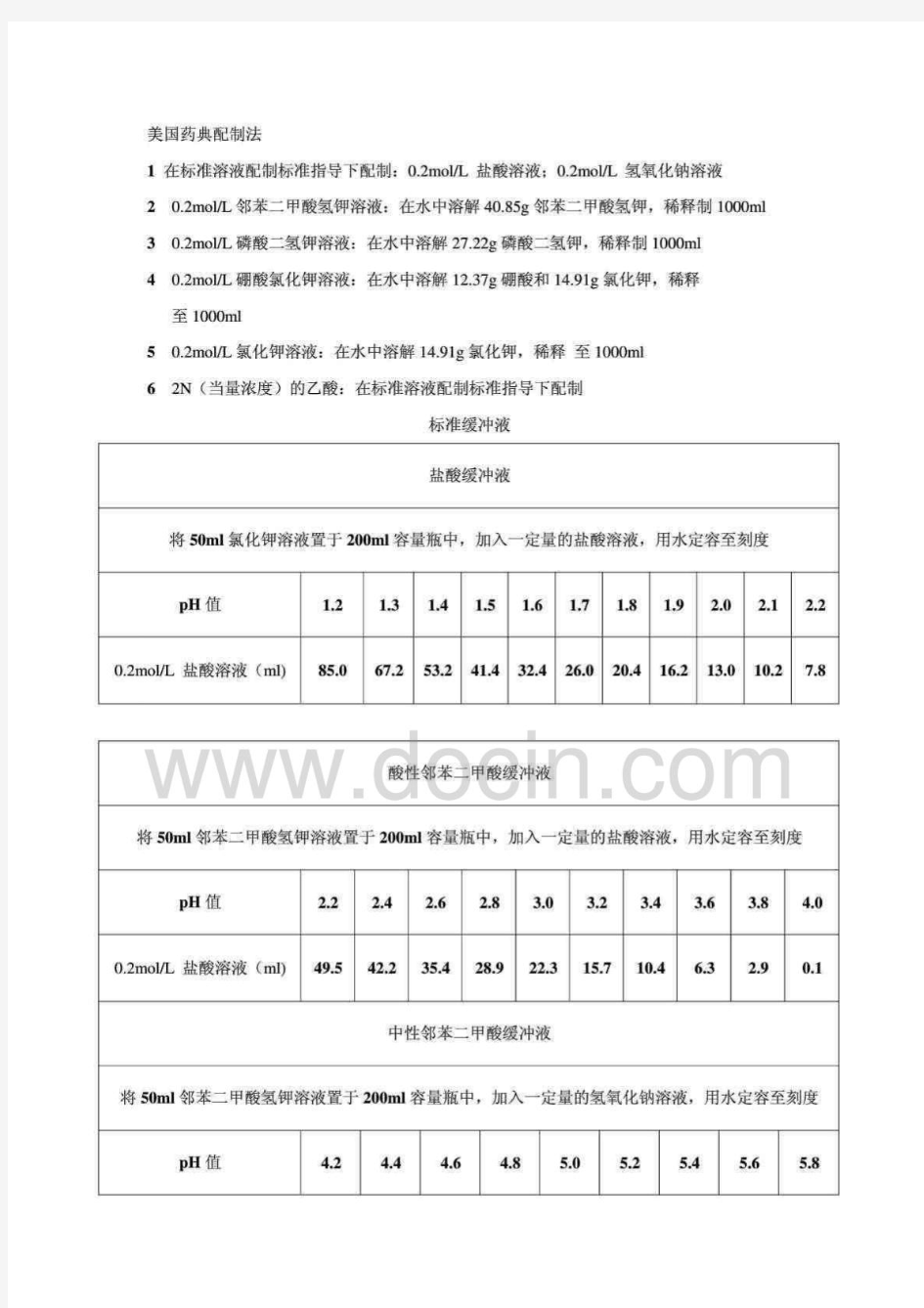
美国药典溶出介质缓冲液的配制
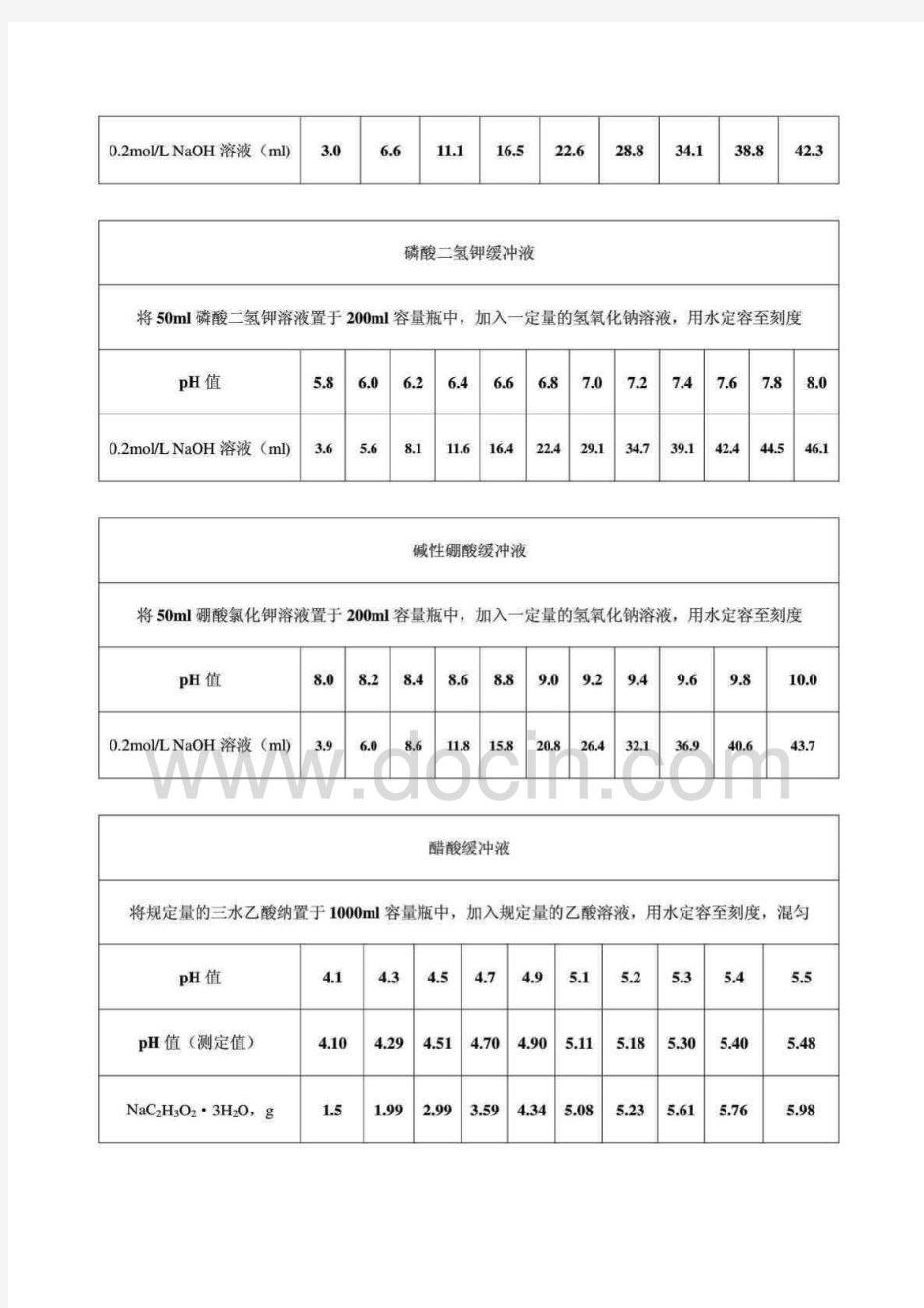
美国药典配制法 1在标准溶液配制标准指导下配制:0.2mol/L 盐酸溶液;0.2mol/L 氢氧化钠溶液 20.2mol/L邻苯二甲酸氢钾溶液:在水中溶解40.85g邻苯二甲酸氢钾,稀释制1000ml 3 0.2mol/L磷酸二氢钾溶液:在水中溶解27.22g磷酸二氢钾,稀释制1000ml 4 0.2mol/L硼酸氯化钾溶液:在水中溶解12.37g硼酸和14.91g氯化钾,稀释 至1000ml 5 0.2mol/L氯化钾溶液:在水中溶解14.91g氯化钾,稀释至1000ml 6 2N(当量浓度)的乙酸:在标准溶液配制标准指导下配制 标准缓冲液 盐酸缓冲液 将50ml氯化钾溶液置于200ml容量瓶中,加入一定量的盐酸溶液,用水定容至刻度 pH值 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 2.0 2.1 2.2 0.2mol/L 盐酸溶液(ml) 85.0 67.2 53.2 41.4 32.4 26.0 20.4 16.2 13.0 10.2 7.8 酸性邻苯二甲酸缓冲液 将50ml邻苯二甲酸氢钾溶液置于200ml容量瓶中,加入一定量的盐酸溶液,用水定容至刻度pH值 2.2 2.4 2.6 2.8 3.0 3.2 3.4 3.6 3.8 4.0 0.2mol/L 盐酸溶液(ml) 49.5 42.2 35.4 28.9 22.3 15.7 10.4 6.3 2.9 0.1 中性邻苯二甲酸缓冲液 将50ml邻苯二甲酸氢钾溶液置于200ml容量瓶中,加入一定量的氢氧化钠溶液,用水定容至刻度pH值 4.2 4.4 4.6 4.8 5.0 5.2 5.4 5.6 5.8
溶出介质的配制
常用溶出介质的配制 表 1 溶出介质 pH值溶出介质 1.0- 2.2 盐酸溶液 3.8- 4.0 醋酸盐缓冲液 4.5- 5.8 醋酸盐或磷酸盐缓冲液 6.8-8.0 磷酸盐缓冲液 上述各溶出介质的组成和配制详述如下: 1.盐酸溶液 取下表中规定量的盐酸,加水稀释至1000ml,摇匀,即得。 表2 盐酸溶液的配制 pH值 1.0 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 2.0 2.1 2.2 盐酸(ml) 9.00 7.65 6.0 4.79 3.73 2.92 2.34 1.84 1.46 1.17 0.92 0.70 2.醋酸盐缓冲液 2mol/L醋酸溶液:取120.0g(114ml)冰醋酸用水稀释至1000ml,即得。取下表中规定物质的取样量,加水溶解并稀释至1000ml,摇匀,即得。 表3 醋酸盐缓冲溶液的配制 pH值 3.8 4.0 4.5 5.5 5.8 醋酸钠(g) 0.67 1.22 2.99 5.98 6.23 2mol/L醋酸溶液(ml)22.6 20.5 14.0 3.0 2.1 3.磷酸盐缓冲液 0.2mol/L磷酸二氢钾溶液:取27.22g磷酸二氢钾,用水溶解并稀释至1000ml。 0.2mol/L氢 氧化钠溶液:取8.00g氢氧化钠,用水溶解并稀释至1000ml。取250ml 0.2mol/L磷酸二氢钾溶液与下表中规定量的0.2mol/L氢氧化钠溶液混合后,再加水稀释至1000ml,摇匀,即得。 表4 磷酸盐缓冲液 pH值 4.5 5.5 5.8 6.0 6.2 6.4 6.6 0.2mol/L氢氧化钠溶液(ml)0 9.0 18.0 28.0 40.5 58.0 82.0 pH值 6.8 7.0 7.2 7.4 7.6 7.8 8.0 0.2mol/L氢氧化钠溶液(ml)118.0 145.5 173.5 195.5 212.0 222.5 230.5 以上为推荐采用的溶出介质配制方法,如有特殊情况,研究者也可根据研究结果采用其他的溶出介质以及相应的配制方法。 PH=1,0.1N HCL溶出介质配置: 9ml浓盐酸(37%)加水稀释成1000ml; PH=4.5醋酸-醋酸钠溶出介质配置:醋酸钠2.99g,加冰醋酸1.596ml。稀释成1000ml; PH=6.8磷酸二氢钾-氢氧化钠溶出介质配置:磷酸二氢钾6.805g,氢氧化钠0.944g。用水稀释成1000ml。 如需配置数升溶出介质,相应增加配置原料的比例。
溶出度指导原则精选版
溶出度指导原则 Document serial number【KKGB-LBS98YT-BS8CB-BSUT-BST108】
普通口服固体制剂溶出曲线?测定与比较指导原则 (2015-11-09 16:15:30) 分类: 普通口服固体制剂溶出曲线测定与比较指导原则 一、概述 为进一步推进仿制药与原研药品质量和疗效一致性评价工作的开展,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)要求,制定本指导原则。 本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。 二、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。 体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。
普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临 床疗效差异的风险。 三、溶出试验方法的建立 溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。可参考有关文献,了解药物的溶解性、渗透性、pKa常数等理化性质,考察溶出装置、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。 (一)溶出仪 溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。 溶出试验推荐使用桨法、篮法,一般桨法选择50~75转/分钟,篮法选择50~100转/分钟。在溶出试验方法建立的过程中,转速的选择推荐由低到高。若转速超出上述规定应提供充分说明。 (二)溶出介质