拟南芥基因克隆的策略与途径

拟南芥基因克隆的策略与途径
拟南芥(Arabidopsis thaliana)是一种模式植物,具有基因组小(125 Mbp)、生长周期短等特点,而且基因组测序
已经完成(The Arabidopsis Genomic Initiative, 2000)。同时,拟南芥属十字花科(Cruciferae),具有高等植物
的一般特点,拟南芥研究中所取得成果很容易用于其它高等植物包括农作物的研究,产生重大的经济效益,特别是十字
花科中还有许多重要的经济作物,与人类的生产生活密切相关,因此目前拟南芥的研究越来越多地受到国际植物学及各
国政府的重视。
基因(gene)是遗传物质的最基本单位,也是所有生命活动的基础。不论要揭示某个基因的功能,还是要改变某个基因的功
能,都必须首先将所要研究的基因克隆出来。特定基因的克隆是整个基因工程或分子生物学的起点。本文就基因克隆的
几种常用方法介绍如下。
1、图位克隆
Map-based cloning, also known as positional cloning, first proposed by Alan Coulson of the University of Cambridge in 1986, Gene isolated by this method is based on functional genes in the genome has a relatively stable loci, in the use of genetic linkage analysis or chromosomal abnormalities of separate groups will queue into the chromosome of a specific location, By constructing high-density molecular linkage map, to find molecular markers tightly linked with the aimed gene, continued to narrow the candidate region and then clone the gene and to clarify its function and biochemical mechanisms.
图位克隆(map-based clonig)又称定位克隆(positoinal cloning),1986年首先由剑桥大学的Alan Coulson提出。用该方法分离基因是根据功能基因在基因组中都有相对较稳定的基因座,在利用分离群体的遗传连锁分析或染色体异常将基因伫到染色体的1个具体位置的基础上,通过构建高密度的分子连锁图,找到与目的基因紧密连锁的分子标记,不断缩小候选区域进而克隆该基因,并阐明其功能和生化机制。
用该方法分离基因是根据目的基因在染色体上的位置进行的,无需预先知道基因的DNA序列,也无需预先知道其表达产物的有关信息。它是通过分析突变位点与已知分子标记的连锁关系来确定突变表型的遗传基础。近几年来随着拟南芥基因组测序工作的完成,各种分子标记的日趋丰富和各种数据库的完善,在拟南芥中克隆一个基因所需要的努力已经大大减少了(图1)。
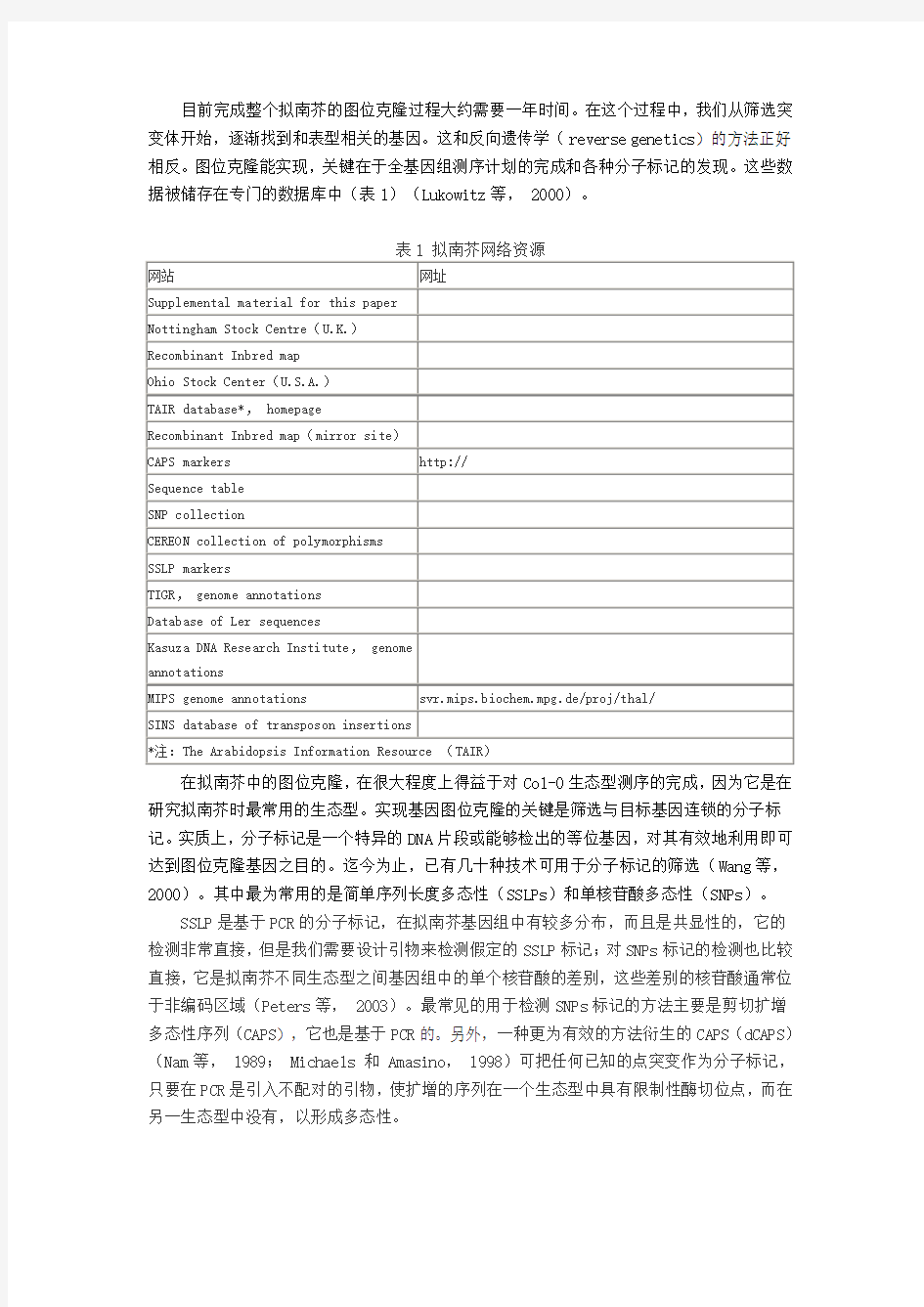
目前完成整个拟南芥的图位克隆过程大约需要一年时间。在这个过程中,我们从筛选突变体开始,逐渐找到和表型相关的基因。这和反向遗传学(reverse genetics)的方法正好相反。图位克隆能实现,关键在于全基因组测序计划的完成和各种分子标记的发现。这些数据被储存在专门的数据库中(表1)(Lukowitz等, 2000)。
在拟南芥中的图位克隆,在很大程度上得益于对Col-0生态型测序的完成,因为它是在研究拟南芥时最常用的生态型。实现基因图位克隆的关键是筛选与目标基因连锁的分子标记。实质上,分子标记是一个特异的DNA片段或能够检出的等位基因,对其有效地利用即可达到图位克隆基因之目的。迄今为止,已有几十种技术可用于分子标记的筛选(Wang等,2000)。其中最为常用的是简单序列长度多态性(SSLPs)和单核苷酸多态性(SNPs)。
SSLP是基于PCR的分子标记,在拟南芥基因组中有较多分布,而且是共显性的,它的检测非常直接,但是我们需要设计引物来检测假定的SSLP标记;对SNPs标记的检测也比较直接,它是拟南芥不同生态型之间基因组中的单个核苷酸的差别,这些差别的核苷酸通常位于非编码区域(Peters等, 2003)。最常见的用于检测SNPs标记的方法主要是剪切扩增多态性序列(CAPS),它也是基于PCR的。另外,一种更为有效的方法衍生的CAPS(dCAPS)(Nam等, 1989; Michaels 和 Amasino, 1998)可把任何已知的点突变作为分子标记,只要在PCR是引入不配对的引物,使扩增的序列在一个生态型中具有限制性酶切位点,而在另一生态型中没有,以形成多态性。
图位克隆法随着相关配套技术(序列数据库、分子标记等)的日渐成熟,许多拟南芥及一些农作物的基因已被成功的克隆(表2)。
本文拟对图位克隆的研究进展做一介绍,以期对植物遗传育种和分子生物学研究有所帮助。
2 图位克隆的一般过程
因为有了拟南芥的基因组序列和高密度的遗传标记,图位克隆过程就变得相对直接。图2例举了一种高效的拟南芥图位克隆方法。从基于Col-0和L er遗传背景的突变体出发,我们有可能在大约一年时间内找出与这个突变相关的基因,这其中主要耗时间的是五个植物(拟南芥)的生长周期(我们假定每个周期为两个月)。
作为作图过程的第一步,突变体植株将和另外一个生态型(Col-0或者L er)的植株杂交。在大多数情况下,用于杂交的突变体植株是作为父本还是母本是没有关系的。然后播种F1代种子。在F1代植物的生长过程中,我们就有可能来对其表现型和基因型进行分析。F1代植物的表型的出现或者消失将显示着我们所研究的突变是显性的还是隐性的。最好通过对一些标记的分析来确认F1代植物是杂合体,而且在杂交过程中我们没有犯错误。当然也有必要确认原来的生态型背景。
图2 图位克隆过程示意图(Jander等, 2002)
F1代植物自交得到F2代种子,大约播种600个个体以进行突变基因的粗定位
(first-pass mapping,图2)。在其生长过程中,我们可确定其表型,大约有150个个体被认为是纯合体(在隐性突变的情况下是纯合突变体,在显性突变的情况下是纯合野生型)。然后从这150个个体的叶子或者其它组织中制备DNA用于基因型分析。起先用分布于拟南芥五条染色体上的25个标记(相邻的两个标记之间大约相距20 cM)进行分析,确定突变基因是和哪个或者哪几个标记是连锁的,然后用三点测交的方法来定义一个包含突变基因的大约20 cM的遗传间隔。一旦这样的一个遗传间隔被定义之后,接下来的工作就是引入新的标记把这个间隔缩小到大约4 cM。一般来说,利用150个F2代个体是在很大程度上能找到这样一个遗传间隔的,距离突变基因最近的两个分子标记将作为侧面标记而用于下面的进一步分析。
下一步我们将播种一个更大的F2代群体用于突变基因的精细定位(fine-resolution mapping,图2)。最终目标是将包含突变基因的遗传间隔缩小到40 Kb甚至更小(这在拟南芥中大约是0.16 cM)。显然用于作图的F2代植物越多,就越能精确地定位突变基因。一般需要3000~4000个F2代植物个体(包括粗定位时的600个F2代植物个体)来精确地定位突变基因。但是也有很多图位克隆过程用了少于3000个F2代植物个体就成功地定位了突变基因(Lukowitz等, 2000)。但是这往往要冒因为作图群体不够大再一次种植F2代植物而延长整个作图过程的时间的风险。
在这个大约4 cM的遗传间隔内找到与突变更紧密连锁的分子标记,一般情况下能在突变两侧找到相距小于40 Kb的两个分子标记。一旦这样的两个分子标记被找到之后,就可以通过测序来找到突变基因。一种有效的方法是设计PCR引物来扩增覆盖这40 Kb的多个重叠的500 bp的片段。将这些片段测序后拼接起来以得到整个40 Kb的序列,然后将它与野生型植物(Col-0或者L er)的序列进行比对,这就可以找到这个区域中的多个基因。从一系
列侯选基因中鉴定基因是定位克隆技术的最后一个关键环节。现在最常用的方法是用含有目标基因的大片段克隆如BAC克隆或YAC克隆去筛选cDNA文库,并查询生物数据信息库,待找出侯选基因后,把这些侯选基因进行下列分析以确定目标基因:(1)精确定位法检查cDNA 是否与目标基因共分离;(2)检查cDNA时空表达特点是否与表型一致;(3)测定cDNA
序列,查询数据库,以了解该基因的功能;(4)筛选突变体文库,找出DNA序列上的变化及与功能的关系;(5)进行功能互补实验,通过转化突变体观察突变体表型是否恢复正常或发生预期的表型变化。功能互补实验是最直接、最终鉴定基因的方法。利用新兴的RNA
干扰(RNAi)也可有效地确定目的基因。
同源克隆:生物的种、属之间编码基因序列的同源性高于非编码区的序列,基于此原理,在其他种属同源基因被克隆的前提下,构建cDNA文库或基因组文库,然后用已知分子高保守序列制备同源探针,经标记后从相应的文库中筛选阳性克隆,并经核酸序列分析鉴定所克隆的基因,当然在没有全同源探针的情况下,可以使用部分同源探针来筛选与探针序列相关但不完全相同的基因。
图位克隆
同源克隆
转座子标签
表达序列标签(expressed sequence tags, EST)是从一个随机选择的cDNA克隆进行5′端和3′端单一次测序获得的短的cDNA部分序列, 代表一个完整基因的一部分。
DbEST,database EST 表达序列标签数据库。
1951年Barbara Mclintock首先在玉米中发现了控制元件,后来命名为转座元件或转座子(transposon)。转座子是基因组中一段可移动的DNA序列,可以通过切割、重新整合等一系列过程从基因组的一个位置“跳跃”到另一个位置。这一元件不仅可用于分析生物遗传进化上分子作用引起的一些现象,还为基因工程和分子生物学研究提供了强有力的工具,可以在不了解基因产物的生化性质和表达
模式的情况下,分离克隆植物基因,即转座子标签(transposon tagging),又称为转座子示踪法。其原理是利用转座子的插入造成基因突变,以转座子序列为基础,从突变株的基因文库中筛选出带有此转座子的克隆,它必定含有与转座子序列相邻的突变基因的部分序列,再利用这部分序列从野生型基因文库中获得完整的基因〔1〕。1984年,用转座子标签法首先在玉米中分离了bronze基因,该基因编码了玉米花色素合成途径的关键酶——UDP-葡萄糖类黄3-O-葡萄糖基转移酶〔2〕。此后还利用转座子标签技术分离了许多植物基因。
转座子可以分为两大类:以DNA-DNA方式转座的转座子和反转录转座子(retrotransposon)。第一类转座子可以通过DNA复制或直接切除两种方式获得可移片段,重新插入基因组DNA中。根据转座的自主性,这类元件又可以分为自主转座元件和非自主转座元件,前者本身能够编码转座酶而进行转座,后者则需在自主元件存在时方可转座,以玉米的Ac/Ds体系为例,Ac(Activator)属于自主元件,Ds(Dissociation)则是非自主元件,必需在Ac元件存在下才能转座〔1〕。第二类转座子又称为返座元(retroposon)〔3〕,是近年新发现的由RNA介导转座的转座元件,在结构和复制上与反转录病毒(retrovirus)类似,只是没有病毒感染必须的env基因,它通过转录合成mRNA,再逆转录合成新的元件整合到基因组中完成转座,每转座1次拷贝数就会增加1份,因此它是目前所知高等植物中数量最大的一类可活动遗传成分。目前共发现了3种类型反转录转座子:
Tyl-copia类,Ty3-gypsy类和LINE(long interspersed nuclear Clements)类转座子,前两类是具有长末端重复的转座子,LINE类转座子没有长末端重复。高等植物中的反转录转座子主要属于Tyl-copia类,分布十分广泛,几乎覆盖了所有高等植物种类〔4〕。
克隆转座子主要有两条途径:其一,利用抗体识别或cDNA探针从野生型植株中获得表达量降低或不稳定基因座的序列,再从突变体中分离得到相应的转座子:其二是根据序列同源性,在基因组的不同位置分离同一家族的转座子成员。目前已经克隆的植物转座子约156种(来自Genbank的报告
4植物基因克隆的策略与方法
4植物基因克隆的策略与方法 基因的克隆确实是利用体外重组技术,将特定的基因和其它DNA顺序插入到载体分子中。基因克隆的要紧目标是识不、分离特异基因并获得基因的完整的全序列,确定染色体定位,阐明基因的生化功能,明确其对特定性状的遗传操纵关系。通过几十年的努力由于植物发育,生理生化,分子遗传等学科的迅速进展,使人们把握了大量有关植物优良性状基因的生物学和遗传学知识,再运用先进的酶学和生物学技术差不多克隆出了与植物抗病、抗虫、抗除草剂、抗逆,育性、高蛋白质及与植物发育有关的许多基因。我们实验室对天麻抗真菌蛋白基因作了功能克隆的研究(舒群芳等,1995;舒群芳等,19 97),为了克隆植物基因也探讨了其它克隆方法,本文论述基因克隆的策略、方法及取得的一些进展。 1功能克隆(functional Cloning) 功能克隆确实是按照性状的差不多生化特性这一功能信息,在鉴定和已知基因的功能后克隆(Collis,1995)。其具体作法是:在纯化相应的编码蛋白后构建cDNA文库或基因组文库,DNA文库中基因的选择按照情形要紧可用二种方法进行,(1)将纯化的蛋白质进行氨基酸测序,据此合成寡核苷酸探针 从cDNA库或基因组文库中选择编码基因,(2)将相应的编码蛋白制成相应抗体探针,从cDNA入载体表达库中选择相应克隆。功能克隆是一种经典的基因克隆策略,专门多基因的分离利用这种策略。 Hain等从葡萄中克隆了两个编码白藜芦醇合成的二苯乙烯合成酶基因(Vst1和Vst2),葡萄中抗菌化合物白藜芦醇的存在,能够提升对灰质葡萄孢(B otrytis cinerce)的抗性,在烟草和其它一些植物中无二苯乙烯合成酶,因此克隆该基因通过转基因后,对有些植物产生对灰质葡萄孢的抗性专门有意义(H ain等,1985)。Kondo等1989年对编码水稻巯基蛋白酶抑制剂的基因组DN A做了克隆和序列分析(Kondo等,1989)。周兆斓等构建了水稻cDNA文库,分离了编码水稻巯基蛋白酶抑制剂的cDNA(周兆斓等,1996)。植物蛋白酶抑制剂是一类天然的抗虫物质,它可抑制摄食害虫对蛋白质的消化,使害虫因 缺乏所需氨基酸而导致非正常发育或死亡。胡天华等人从烟草中分离出流行于我国的黄瓜花叶病毒(Cucumber Mosaic virus)(CMV),并克隆了编码该
整个基因克隆实验流程完整
一、组织总RNA的提取 相关试剂:T rizol;氯仿;苯酚;异丙醇;75%乙醇;RNase-free水 相关仪器:制冰机;液氮&研钵/生物样品研磨仪;高速离心机;移液器(1ml、200μl、100μl/50μl);涡旋振荡仪;恒温金属浴。 相关耗材:解剖工具,冰盒,离心管,离心管架,吸头(1ml,200μl/300μl),一次性手套,实验手套。 实验步骤 1.取暂养草鱼,冰上放置一段时间,然后解剖,剪取肠道50~100mg,放入研钵中,加入 液氮迅速研磨,然后加入1ml 预冷TRIzol试剂,充分研磨至无颗粒物存在。 2.转移到离心管中,室温放置5min,使细胞充分裂解; 3.按1ml Trizol加入200μl氯仿,盖上盖子,迅速充分摇匀15s,然后室温放置3min; 4.4℃,,12000g 离心15min; 此时混合物分为三层,下层红色的苯酚氯仿层,中间层和上层无色水相;RNA存在于无色水相中; 5.小心吸取上清液,千万不要吸取中间界面,否则有DNA污染;转移至一个新的离心管, 加入等体积的异丙醇,轻轻混匀; 6.室温放置10min;4℃,,12000g 离心10min; 7.弃上清,加入1ml 75%乙醇洗涤;涡旋,悬浮沉淀;4℃,,12000g 离心5min; 8.弃上清;可以再次用75%乙醇洗涤沉淀; 9.弃上清;用移液器轻轻吸取管壁或管底的残余乙醇,注意不要吸取沉淀;室温放置5min 晾干沉淀;(RNA样品不要过于干燥,否则极难溶解) 10.沉淀中加入30μl RNase-free水,轻弹管壁,使RNA溶解。 RNA质量检测 相关试剂:溴酚蓝,TEB/TAE电泳缓冲液,溴乙锭(EB) 相关仪器:(超微量分光光度计,移液器(2.5μl 或2μl 规格,10μl规格),电子天平,电泳仪,电泳槽,凝胶成像仪,微波炉,制冰机) 相关耗材:(无菌无绒纸,吸头,离心管架,PCR管,PCR管架,锥形瓶,烧杯,一次性手套,实验手套,冰盒) (1)RNA纯度的检测:测定其OD260和OD280的值,根据其OD260/ OD280的比值,当其比值在1.9~2.1之间,说明提取的总RNA纯度比较高,没有蛋白质和基因组的污染。 (2)RNA完整性的检测:取2μlRNA,与2μl溴酚蓝混匀,用1%的琼脂糖进行凝胶电泳,20min后,在凝胶成像系统中观察效果。当28S与18S条带清晰,且亮度比大约是2:1时,5S条带若隐若现,而且没有其它条带时,说明完整性不错,可以用于下游逆转录实验。
基因克隆、假病毒操作步骤
实验名称:基因克隆 实验器材:荧光定量PCR仪、摇床、离心机、生工PCR产物纯化试剂盒、恒温加热器、 NEB连接体系、灭菌纯水、JM109感受态、冰、LB培养基、酒精灯、涂棒、氨苄、氨苄抗性平板、甘油等; 操作步骤: 1、可通过PCR进行拼接获得目的基因的,过柱纯化(生工试剂盒根据说明书进行纯化, 在最后一步的洗脱可以用预热的灭菌纯水洗脱,在加灭菌纯水洗脱的时候一定要加在纯化柱子的膜中间); 2、选择合适的载体(EZ-T)用连接酶进行连接,NEB体系,16℃过夜连接 T4lages 1.0 10×T4buffer 2.0 EZ-T 1.0 目的基因8.0 DdH2O 8.0 _________ 20ul 3、取100μl摇匀后的JM109感受态细胞悬浮液(如是冷冻保存液,则需化冻后马上进行下 面的操作),加入10μl连接产物,轻轻摇匀,冰上放置30min后,于42度水浴中保温90s,然后迅速在冰上冷却2min; 4、加入500μl LB液体培养基,混匀于37℃振荡培养45min使受体菌恢复正常生长状态并 使转化体产生抗药性; 5、将恢复培养的菌体5000rpm离心3min,移去上层LB培养基,用余下的200μl重悬菌体, 并用灭菌玻璃推子(酒精灯上烧后冷却),均匀涂布于琼脂凝胶表面(氨苄抗性),37℃倒置培养12~16小时; 6、挑取多个单克隆菌落分别接种到1ml含有抗生素(氨苄)的LB液体培养基中,37℃振 荡培养3h; 7、培养1-2小时即可以利用PCR(定量或定性)进行鉴定; 8、选取初步鉴定阳性的菌液送测序,测序正确后甘油保存(甘油的浓度为30%-50%),充 分混匀,-80℃保存;
基因克隆及转基因方法
基因克隆及转基因 一、基因克隆及转基因过程 1、设计引物 软件是https://www.360docs.net/doc/5d16308285.html,sergene.v7.1,用到里面的PrimerSelect和EditSeq。 一般原则:1、长度:18-25; 2、GC含量:40-60%,正反向引物相差不要大于5%; 3、Tm值:55以上(到65),实在不行50以上也可以,正反向引物相差不要大 于5; 4、3’端结尾最好是GC,其次是T,不要A; 5、正反向引物连续配对数小于4; 6、在NCBI上的Primer Blast上看引物特异性如何; (如果克隆的话不能满足条件也没办法。) 不是必须条件,但可以考虑:多个基因设计引物时,可尽量使Tm值相似,方便PCR。 步骤: 一、打开PrimerSelect和EditSeq。 二、在EditSeq中输入你的序列。 引物有一对F和R 1、对于F是从5’到3’,在序列的前部分选择长度为18-25bp的碱基,如果你是要验证就随便选,如果你是要克隆就在最开始选,不符合原则就只能在你选的后边增或减碱基。 2、将选择的F引物输入到PrimerSelect中,在File中选择Enter New Primer,复制,OK,然后可以看到引物的情况,看看长度、Tm、GC含量是不是符合标准,不符合就继续选。 3、对于R是从3’到5’,选中序列,在EditSeq的Goodies中选择第一个“反向互补”,此时序列已反向互补,按照前面F的方法搜索R的引物。、 4、注意你想要的目的带的大小,比如序列是1000bp,你想PCR出来800大小的目的带,那就要看看F和R之间的长度在你想要的范围内。可以将R反向互补,在正向的序列中搜索R在的位置,就是在EditSeq中选择Search,点击第一个Find,开始搜寻。 5、搜索完引物在PrimerSelec中的Report中选择前两个查看二聚体情况。 6、在NCBI上的Primer Blast上看引物特异性如何。 7、因为是克隆,所以引物要有酶切位点,酶切位点的加入主要考虑所用到的表达载体,在NEBcutter网站中输入总序列查看可用的酶切位点。在引物上游加入酶切位点,注意加入时载体的表达的方向,前面的酶切位点在引物F上,后面的酶切位点在引物R上。一般在引物上游还要加上两个保护碱基。 2、提取醋栗DNA 3、PCR扩增与目的基因回收 PCR先找合适的退火温度,找到后回收时就可以多PCR几管,一般我们用20ul的体系,PCR5管就可以回收,就是琼脂糖凝胶回收,将目的基因用刀片切下来,用试剂盒回收。回收完可以再跑电泳检测一遍。 PCR: 20ul体系:灭菌水13.8ul,若模板为质粒灭菌水14.3ul; 2.5mMdNTP2.0ul;
南方医科大学分生实验-绿色荧光蛋白(EGFP)的基因克隆
绿色荧光蛋白(EGFP)的基因克隆 南方医科大学学院 摘要 本实验旨在学习基因克隆并检验,绿色荧光蛋白基因转化入宿主细胞后很稳定,对多数宿主的生理无影响,是常用的报道基因,便于实验。本实验通过将含有目的基因GFP的pEGFP-N1质粒和pMD18-T载体进行酶切、电泳、回收、连接、转入、筛选之后,把GFP基因成功导入到大肠杆菌DH5α(克隆菌)中,从而实现荧光蛋白基因的克隆和表达。 关键词:绿色荧光蛋白克隆表达 实验名称绿色荧光蛋白的基因克隆 2015- ~ 实验日期 实验地点 2015- 合作者指导老师 评分教师签名批改日期 一、实验目的 1.学习使用限制性内切酶进行DNA酶切的原理和方法。 2.学习掌握琼脂糖凝胶电泳的基本原理和操作方法。 3.掌握PCR技术原理和PCR仪的操作方法。 4.学习PCR产物的TA克隆的基本原理和操作步骤。 5.了解和掌握大肠杆菌的制备方法的基本原理和操作要点以及DNA转化大肠杆菌的原理和方 法。
6.掌握双酶切法鉴定重组DNA的基本原理和操作步骤,以及菌落PCR鉴定重组DNA的基本原 理和方法。 7.掌握IPTG诱导GFP基因表达的基本原理和操作步骤 二、实验原理 1.pEGFP-N1质粒 2.T载体
三、材料与方法: 1.实验材料: 质粒:pEGFP-N1 T载体:pUCm-T 菌种:DH5(克隆菌) PCR引物: F——GGCATATGGTGAGCAAGGGCGA R——CGGGATCCCTTGTACAGCTCGTC Tm=56 实验试剂: 即用型蓝白T载体(pMD18-T vector cloning kit) 快速DNA连接试剂盒 限制性内切酶:EcoR I(Fermentas) Axygen质粒提取试剂盒 抗生素:氨苄青霉素(Amp)、卡那霉素(Kan) X-gal、IPTG等 实验仪器: 超净工作台,恒温摇床,高压灭菌锅,恒温培养箱,台式高速离心机,大容量冷冻离心机,PCR仪,紫外分光光度计,水平电泳槽,垂直电泳槽,电泳仪,凝胶成像系统,制冰机、超低温冰箱等 2.方法 分离目的基因→限制酶切割目的基因与载体→连接重组体→转入受体细胞→筛选重组体、转化子 四、实验具体流程 1.获取外源基因 1)碱裂解法提取质粒 使用Axygen质粒提取试剂盒
植物基因的克隆|植物基因克隆的基本步骤
植物基因的克隆 08医用二班姚桂鹏0807508245 简介 克隆(clone)是指一个细胞或一个生物个体无性繁殖所产生的后代群体。通常所说的基因克隆是指基于大肠埃希菌的DNA片段(或基因)的扩增,主要过程包括目标DNA的获取、重组载体的构建、受体细胞的转化以及重组细胞的筛选和繁殖等。本文主要介绍植物基因的特点、基因克隆的载体、基因克隆的工具酶、基因克隆的策略以及植物目的基因的分离克隆方法等内容。 关键词 植物基因基因克隆载体工具酶克隆策略分离克隆方法 Plant gene cloning Introduction Cloning (clone) refers to a cell or an individual organisms asexual reproduction produced offspring. Usually said cloning genes means
based on escherichia coli segment of DNA (or genes), including the main course target DNA, restructuring of the carrier, transformation of receptor cells and reorganization of screening and reproductive cells. This paper mainly introduces the characteristics of plant gene and gene cloning and carrier, gene clone tool enzyme, gene cloning and plant gene strategy of separation cloning method, etc. Keywords Plant gene cloning tool enzyme gene cloning vector method of separation of cloning strategy 一、植物基因的结构和功能 基因(gene)是核酸分子中包含了遗传信息的遗传单位。一般来说,植物基因都可分为转录区和非转录的调控区两部分。 (一)植物基因的启动子 启动子(promoter)是指在位于结构基因上游决定基因转录起始的区域,植物积阴德启动子包括三个较重要的区域,一时转录起始位点,而是转录起始位点上游25~40bp的区域,三是转录起始位点上游-75bp处或更远些的区域。 (二)植物基因的增强子序列
植物基因克隆的策略与方法
植物基因克隆的策略与方法 基因的克隆就是利用体外重组技术,将特定的基因和其它DNA顺序插入到载体分子中。基因克隆的主要目标是识别、分离特异基因并获得基因的完整的全序列,确定染色体定位,阐明基因的生化功能,明确其对特定性状的遗传控制关系。通过几十年的努力由于植物发育,生理生化,分子遗传等学科的迅速发展,使人们掌握了大量有关植物优良性状基因的生物学和遗传学知识,再运用先进的酶学和生物学技术已经克隆出了与植物抗病、抗虫、抗除草剂、抗逆,育性、高蛋白质及与植物发育有关的许多基因。我们实验室对天麻抗真菌蛋白基因作了功能克隆的研究(舒群芳等,1995;舒群芳等,1997),为了克隆植物基因也探讨了其它克隆方法,本文论述基因克隆的策略、方法及取得的一些进展。 1 功能克隆(functional Cloning) 功能克隆就是根据性状的基本生化特性这一功能信息,在鉴定和已知基因的功能后克隆(Collis,1995)。其具体作法是:在纯化相应的编码蛋白后构建cDNA文库或基因组文库,DNA文库中基因的筛选根据情况主要可用二种办法进行,(1)将纯化的蛋白质进行氨基酸测序,据此合成寡核苷酸探针从cDNA库或基因组文库中筛选编码基因,(2)将相应的编码蛋白制成相应抗体探针,从cDNA入载体表达库中筛选相应克隆。功能克隆是一种经典的基因克隆策略,很多基因的分离利用这种策略。 Hain等从葡萄中克隆了两个编码白藜芦醇合成的二苯乙烯合成酶基因(Vst1和Vst2),葡萄中抗菌化合物白藜芦醇的存在,可以提高对灰质葡萄孢(Botrytis cinerce)的抗性,在烟草和其它一些植物中无二苯乙烯合成酶,因此
基因克隆载体上的各种常用蛋白标签
基因克隆载体上的各种常用蛋白标签 蛋白标签(proteintag)是指利用DNA体外重组技术,与目的蛋白一起融合表达的一种多肽或者蛋白,以便于目的蛋白的表达、检测、示踪和纯化等。随着技术的不断发展,研究人员相继开发出了具有各种不同功能的蛋白标签。目前,这些蛋白标签已在基础研究和商业化产品生产等方面得到了广泛的应用。 美国GeneCopoeia(复能基因)为客户提供50多种蛋白标签,可以满足客户的不同需求,包括各种最新型的标签,如:SNAP-Tag?、Halo Tag?、AviTag?、Sumo等;也提供齐全的各种常用标签,如eGFP、His、Flag等等标签。 以下是部分蛋白标签的特性介绍,更加详细的介绍可在查询产品的结果列表里面看到各种推荐的蛋白标签和载体。 TrxHIS His6是指六个组氨酸残基组成的融合标签,可插入在目的蛋白的C末端或N末端。当某一个标签的使用,一是能构成表位利于纯化和检测;二是构成独特的结构特征(结合配体)利于纯化。组氨酸残基侧链与固态的镍有强烈的吸引力,可用于固定化金属螯合层析(IMAC),对重组蛋白进行分离纯化。使用His-tag有下面优点: 标签的量小,只有~0.84KD,而GST和蛋白A分别为~26KD和~30KD,一般不影响目标蛋白的功能; His标签融合蛋白可以在非离子型表面活性剂存在的条件下或变性条件下纯化,前者在纯化疏水性强的蛋白得到应用,后者在纯化包涵体蛋白时特别有用,用高浓度的变性剂溶解后通过金属螯和去除杂蛋白,使复性不受其它蛋白的干扰,或进行金属螯和亲和层析复性; His标签融合蛋白也被用于蛋白质-蛋白质、蛋白质-DNA相互作用研究; His标签免疫原性相对较低,可将纯化的蛋白直接注射动物进行免疫并制备抗体。 可应用于多种表达系统,纯化的条件温和; 可以和其它的亲和标签一起构建双亲和标签。 Flag标签蛋白 Flag标签蛋白为编码8个氨基酸的亲水性多肽(DYKDDDDK),同时载体中构建的Kozak序列使得带有FLAG的融合蛋白在真核表达系统中表达效率更高。FLAG作为标签蛋白,其融合表达目的蛋白后具有以下优点: FLAG作为融合表达标签,其通常不会与目的蛋白相互作用并且通常不会影响目的蛋白的功能、性质,这样就有利用研究人员对融合蛋白进行下游研究。 融合FLAG的目的蛋白,可以直接通过FLAG进行亲和层析,此层析为非变性纯化,可以纯化有活性的融合蛋白,并且纯化效率高。 FLAG作为标签蛋白,其可以被抗FLAG的抗体识别,这样就方便通过Western Blot、ELISA等方法对含有FLAG的融合蛋白进行检测、鉴定。
拟南芥基因克隆的策略与途径
拟南芥基因克隆的策略与途径 拟南芥(Arabidopsis thaliana)是一种模式植物,具有基因组小(125 Mbp)、生长周期短等特点,而且基因组测序 已经完成(The Arabidopsis Genomic Initiative, 2000)。同时,拟南芥属十字花科(Cruciferae),具有高等植物 的一般特点,拟南芥研究中所取得成果很容易用于其它高等植物包括农作物的研究,产生重大的经济效益,特别是十字 花科中还有许多重要的经济作物,与人类的生产生活密切相关,因此目前拟南芥的研究越来越多地受到国际植物学及各 国政府的重视。 基因(gene)是遗传物质的最基本单位,也是所有生命活动的基础。不论要揭示某个基因的功能,还是要改变某个基因的功 能,都必须首先将所要研究的基因克隆出来。特定基因的克隆是整个基因工程或分子生物学的起点。本文就基因克隆的 几种常用方法介绍如下。 1、图位克隆 Map-based cloning, also known as positional cloning, first proposed by Alan Coulson of the University of Cambridge in 1986, Gene isolated by this method is based on functional genes in the genome has a relatively stable loci, in the use of genetic linkage analysis or chromosomal abnormalities of separate groups will queue into the chromosome of a specific location, By constructing high-density molecular linkage map, to find molecular markers tightly linked with the aimed gene, continued to narrow the candidate region and then clone the gene and to clarify its function and biochemical mechanisms. 图位克隆(map-based clonig)又称定位克隆(positoinal cloning),1986年首先由剑桥大学的Alan Coulson提出。用该方法分离基因是根据功能基因在基因组中都有相对较稳定的基因座,在利用分离群体的遗传连锁分析或染色体异常将基因伫到染色体的1个具体位置的基础上,通过构建高密度的分子连锁图,找到与目的基因紧密连锁的分子标记,不断缩小候选区域进而克隆该基因,并阐明其功能和生化机制。 用该方法分离基因是根据目的基因在染色体上的位置进行的,无需预先知道基因的DNA序列,也无需预先知道其表达产物的有关信息。它是通过分析突变位点与已知分子标记的连锁关系来确定突变表型的遗传基础。近几年来随着拟南芥基因组测序工作的完成,各种分子标记的日趋丰富和各种数据库的完善,在拟南芥中克隆一个基因所需要的努力已经大大减少了(图1)。
基因克隆步骤
实验一大肠杆菌感受态细胞的制备及转化 [实验原理](供参考,试剂盒的Solution SS成分未知) 细菌处于容易吸收外源DNA的状态叫感受态。转化是指质粒DNA 或以它为载体构建的重组子导入细菌的过程。其原理是:在0℃下的CaCl2低渗溶液中,细菌细胞膨胀成球形。转化缓冲液中的DNA形成不易被DNA酶所降解的羟基—钙磷酸复合物,此复合物粘附于细菌细胞表面。42℃短时间热处理(热休克),可以促进细胞吸收DNA复合物。将处理后的细菌放置在非选择性培养液中保温一段时间,促使在转化过程中获得的新的表型(如Amp抗性) 得到表达。然后再涂布于含有氨苄青霉素的选择性平板上,37℃培养过夜,这样即可得到转化菌落。[仪器、材料与试剂] (一)仪器1.小型高速离心机2.恒温摇床3.恒温箱4.‐20℃冰箱5.恒温水浴器 (二)材料1.氨苄青霉素2.大肠杆菌DH5a3.pUC194.1.5mL 离心管5.枪头、枪6.试管、培养皿 (三)试剂1.快速感受态细菌制备试剂盒(申能博彩公司产品)2.LB 培养液在950mL去离子水中加入:胰蛋白胨(tryptone) 10g酵母提取物(yeast extract) 5g NaCl 10g 摇动容器直至溶质完全溶解,用Na0H调节pH至7.0,加入去离子水至总体积为1L,121℃湿热灭菌20min。 3.氨苄青霉素(Amp),用无菌水配制成100mg/mL 溶液,置‐20℃冰箱保存。 [实验步骤]
1.从大肠杆菌DH5a平板上挑取一个单菌落接于2mL LB培养液的试管中,37℃振荡培养过夜。 2.取50mL菌液转接到一个含有5mL LB培养液锥形瓶中,37℃振荡培养2小时。以下步骤按修改后的试剂盒说明书进行。 3.用灭菌的枪头取0.5mL的大肠杆菌培养物于1.5mL灭菌离心管中,冰上放置3分钟后,加入0.5mL预冷的Solution SS。在冰上小心地用1mL 枪头将细胞悬浮起来。注意:1mL的取液器设定在500mL。悬浮细胞要轻,防止细胞进入枪内。 4.将上述细胞分装于1.5mL离心管(离心管要在放在冰上预冷) 中,每管0.1mL。细胞可以立即使用或储存。 5.将感受态细胞迅速转移到‐20℃或更低的低温冰中。注意:在转移过程中要防止温度升高,解决的办法之一是在塑料袋里装上低温冰块,将细胞迅速转移到塑料里,将整个塑料袋放到低温冰箱内。 转化:1.新鲜制备的或‐20℃下保存的100mL感受态细胞,置于冰上,完全解冰后轻轻地将细胞均匀悬浮。 2.加入5mL pUC19质粒,DNA浓度为10pg/mL,轻轻混匀。 3.冰上放置30分钟。 4.42℃水浴热激60秒。 5.冰上放置2分钟。 6.加400mL LB培养液,37℃ 250转/分振荡培养30分钟。 7.室温下4000rpm离心5分钟,用枪头吸掉400mL上清液,用剩余的培养液将细胞悬浮。
植物基因克隆实验指导
植物基因克隆实验规则 一、植物基因克隆实验课的目标 根据基因克隆实验操作的整体性和连贯性特点, 将该实验设计为综合性实验课程,实验内容设计上完全抛弃了原来分散的、孤立的单纯学习某一实验技术的缺陷, 将单个实验综合为系统的、连贯的系列型大实验,注重科研成果在教学中的应用,我们从以往的科研项目中选取了部分研究内容用于学生的综合性实验教学,这是基于教学实验与实际科学研究实验之间的新的实验教学模式。 整套实验围绕洋甘菊倍半萜生物合成途径中关键酶基因HMGR的克隆这一研究课题进 行操作, 设计的实验内容具有极强的连续性和综合性,让学生在独立实践操作中学习基因克隆的基本研究方法和体会科学研究的严密逻辑和培养科研理念。 我们将实验内容设置为8个部分, 实验内容前后衔接紧密, 环环相扣, 不可分割, 前一个实验的结果是下一个实验的材料。该课程使学生获得了整个类似科研实践过程的训练和体验, 学习了从事科研工作的基本功, 对完成自己的毕业论文及将来从事生命科学研究奠定了科 研基础。 二、实验的进行程序和要求 1、预习学生在课前应认真预习实验指导以及教材有关章节,必须对该次实验的目的要求、实验内容、基本原理和操作方法有一定的了解。 2、讲解教师对该实验内容的安排及注意事项进行讲解,让学生有充分的时间按实验指导的要求进行独立操作与观察。 3、独立操作与观察除个别实验分组进行外,一般由学生个人独立进行操作和观察。在实验中要按实验指导认真操作,仔细观察,作好记录。有关基本技能的训练,要按操作程序反复练习,以达到一定的熟练程度。
4、演示每次的实验都备有演示内容,其目的是帮助学生了解某些实验中的难点,扩大在实验课有限时间内获得更多感性知识的机会。 5、作业实验报告参照硕士毕业论文的格式写,必须强调科学性,实事求是地记录、分析、综合。在实验结束时呈交。 6、小结每次实验结束后,由师生共同小结本次实验的主要收获及今后应注意的问题。 三、实验规则和注意事项 1、每次上课前,必须认真阅读实验指导,明确本次实验的目的要求、实验原理和注意事项,熟悉实验内容、方法和步骤。 2、上实验课时必须携带实验指导、记录本及文具等。进入实验室要按规定座位入座。 3、实验时要遵守纪律,听从教师指导,保持肃静。有问题时举手提问,严禁彼此谈笑喧或随意走动,也不得私自进行其他活动。 4、实验时要遵守实验操作规程,严格按照教师的安排和实验指导的要求进行。操作观察要认真仔细,边做、边看、边想,认真做好实验记录。 5、要爱护仪器和器材设备,注意节约实验材料、药品和水电。如有损坏器材应立即报告并主动登记、说明情况。 6、实验结束后,应清理实验台面,认真清理好仪器、药品及其他用品,放回原处,放好凳子,方可离开实验室。值日生要负责清扫地面,收拾实验用品,处理垃圾,关好水、电、门窗后再离开。
整个基因克隆实验流程(完整)
整个基因克隆实验流程(完 整) -标准化文件发布号:(9456-EUATWK-MWUB-WUNN-INNUL-DDQTY-KII
一、组织总RNA的提取 相关试剂:T rizol;氯仿;苯酚;异丙醇;75%乙醇;RNase-free水 相关仪器:制冰机;液氮&研钵/生物样品研磨仪;高速离心机;移液器(1ml、200μl、100μl/50μl);涡旋振荡仪;恒温金属浴。 相关耗材:解剖工具,冰盒,离心管,离心管架,吸头(1ml,200μl/300μl),一次性手套,实验手套。 实验步骤 1.取暂养草鱼,冰上放置一段时间,然后解剖,剪取肠道50~100mg,放入研 钵中,加入液氮迅速研磨,然后加入1ml 预冷TRIzol试剂,充分研磨至无颗粒物存在。 2.转移到离心管中,室温放置5min,使细胞充分裂解; 3.按1ml Trizol加入200μl氯仿,盖上盖子,迅速充分摇匀15s,然后室温放置 3min; 4.4℃,,12000g 离心15min; 此时混合物分为三层,下层红色的苯酚氯仿层,中间层和上层无色水相; RNA存在于无色水相中; 5.小心吸取上清液,千万不要吸取中间界面,否则有DNA污染;转移至一个 新的离心管,加入等体积的异丙醇,轻轻混匀; 6.室温放置10min;4℃,,12000g 离心10min; 7.弃上清,加入1ml 75%乙醇洗涤;涡旋,悬浮沉淀;4℃,,12000g 离心 5min; 8.弃上清;可以再次用75%乙醇洗涤沉淀; 9.弃上清;用移液器轻轻吸取管壁或管底的残余乙醇,注意不要吸取沉淀;室 温放置5min晾干沉淀;(RNA样品不要过于干燥,否则极难溶解) 10.沉淀中加入30μl RNase-free水,轻弹管壁,使RNA溶解。 RNA质量检测 相关试剂:溴酚蓝, TEB/TAE电泳缓冲液,溴乙锭(EB) 相关仪器:(超微量分光光度计,移液器(2.5μl 或2μl 规格,10μl规格),电子天平,电泳仪,电泳槽,凝胶成像仪,微波炉,制冰机) 相关耗材:(无菌无绒纸,吸头,离心管架,PCR管,PCR管架,锥形瓶,烧杯,一次性手套,实验手套,冰盒) (1)RNA纯度的检测:测定其OD260和OD280的值,根据其OD260/ OD280的比值,当其比值在1.9~2.1之间,说明提取的总RNA纯度比较高,没有蛋白质和基因组的污染。 (2)RNA完整性的检测:取2μlRNA,与2μl溴酚蓝混匀,用1%的琼脂糖进行凝胶电泳,20min后,在凝胶成像系统中观察效果。当28S与18S条带清晰,
基因克隆的几种常见方法
基因克隆得几种常见方法 基因(gene)就是遗传物质得最基本单位,也就是所有生命活动得基础。不论要揭示某个基因得功能,还就是要改变某个基因得功能,都必须首先将所要研究得基因克隆出来。特定基因得克隆就是整个基因工程或分子生物学得起点。本文就基因克隆得几种常用方法介绍如下。 1 根据已知序列克隆基因 对已知序列得基因克隆就是基因克隆方法中最为简便得一种。获取基因序列多从文献中查取,即将别人报道得基因序列直接作为自己克隆得依据。现在国际上公开发行得杂志一般都不登载整个基因序列,而要求作者在投稿之前将文章中所涉及得基因序列在基因库中注册,拟发表得文章中仅提供该基因在基因库中得注册号(accession number),以便别人参考与查询。目前,世界上主要得基因库有1)EMBL,为设在欧洲分子生物学实验室得基因库,其网上地址为; (2)Genbank,为设在美国国家卫生研究院(NIH)得基因库,其网上地址为;(3)Swissport与TREMBL,Swissport就是一蛋白质序列库,其所含序列得准确度比较高,而TREMBL只含有从EMBL库中翻译过来得序列。目前,以Genbank得应用最频繁。这些基因库就是相互联系得,在Genbank注册得基因序列,也可能在Swissport注册。要克隆某个基因可首先通过Internet查询一下该基因或相关基因就是否已经在基因库中注存。查询所有基因文库都就是免费得,因而极易将所感兴趣得基因从库中拿出来,根据整个基因序列设计特异得引物,通过PCR从基因组中克隆该基因,也可以通过RT-PCR克隆cDNA。值得注意得就是,由于物种与分离株之间得差异,为了保证PCR扩增得准确性,有必要采用两步扩增法,即nested PCR。 根据蛋白质序列也可以将编码该蛋白质得基因扩增出来。在基因文库中注册得蛋白质序列都可以找到相应得DNA或cDNA序列。如蛋白质序列就是自己测定得,那么需要设计至少1对简并引物(degenerated primer),从cDNA文库中克隆该基因。以这种方法克隆得基因必须做序列测定才能鉴别所扩增产物得特异性。 另外,在基因克隆之后,如还要进一步做表达研究,所使用得PCR酶最好不用Taq DNA聚合酶,而采用其她有自我检测(reading proof)功能得酶,如pfu。这样可以避免由于扩增过程中出现得点突变或终止密码子而导致整个研究结论得错误。 2根据已知探针克隆基因 这也就是基因克隆得一种较直接得方法。首先将探针作放射性或非放射性标记,再将其与用不同内切酶处理得基因组DNA杂交,最后将所识别得片段从胶中切下来,克隆到特定得载体(质粒、噬菌体或病毒)中作序列测定或功能分析。这种方法不但可以将基因克隆出来,还能同时观察该基因在基因组中得拷贝数。
山羊基因克隆实验方案设计
山羊基因克隆实验方案设计 第五组 1. 提取DNA 2. DNA检测(电泳) 3. 目的DNA片段基因扩增(PCR) 4. DNA检测(电泳) 5. 目的基因片段与载体连接 6. 大肠杆菌感受态细胞的制备 7. 导入受体细胞与蓝白斑筛选 8. 阳性克隆鉴定 9. DNA测序 一、DNA提取 【实验目的】 指导DNA提取原理(羔羊片),熟悉DNA提取步骤。 【实验原理】 组织细胞被PK消化后,经过萃取漂洗获得纯净DNA。 【实验步骤】 1. <30 mg组织,用眼科剪剪碎,加入200 TL buffer+20 μL PK,55℃水浴1-3 h。 2. 加入220 μL buffer BL,振荡15 s,70 ℃放置10 min,溶液变清亮。 3. 10000 r离心2 min,移上清液至新离心管。 4. 加220 μL无水乙醇,充分振荡15 s,可能会出现絮状沉淀。 5. 将溶液和絮状物加入到吸附柱中,12000 r离心30 s,弃废液。 6. 向吸附柱加入500 μL WB,12000 r离心30 s,弃废液。 7. 向吸附柱加入500 μL wash buffer,12000 r 离心30 s,弃废液,空滤一次。 8. 将吸附柱转入干净离心管,加入50 μL TE,放置5 min,12000 r离心2 min,备检。 二、DNA电泳检测 【实验原理】 琼脂糖凝胶电泳技术是分离、鉴定和提纯DNA片断的有效方法。凝胶分辨率决定于使用材料的浓度,并由此决定凝胶的孔径。琼脂糖凝胶可分辩0.1~6.0kb的双链DNA片段。琼
脂糖凝胶电泳是一个电场作用。它首先利用琼脂糖的分子筛效应,此外,在弱碱性条件下,DNA分子带负电荷,从负极向正极移动。根据DNA分子大小、结构及所带电荷的不同,它们以不同的速率通过介质运动而相互分离。借助溴化乙锭(EB)能与双链DNA结合的作用,利用EB染色,并通过紫外线激发即可观察被分离DNA片段的位置。 【仪器与试剂】 琼脂糖、10×TAE电泳缓冲液(40mMTris,20mMNaAc,1mMEDTAPH8.0)、载体缓冲液(0.25%溴酚蓝,30%甘油)、溴化乙锭水溶液(10mg/ml)、凝胶槽、电泳仪 【实验步骤】 1.取琼脂糖0.9g,加入100ml1xTAE电泳缓冲液于250ml烧瓶中,100℃加热溶解。 2.平衡凝胶槽,放好两侧挡板,调节好梳子与底板的距离(一般高出底板0.5~1mm)。 3.铺板:在溶解好的凝胶中加入终浓度为0.5ug/ml的溴化乙锭水溶液,轻轻混匀,待冷至50℃左右倒入凝胶槽,胶厚一般为5~8mm。 4.待胶彻底凝固后,去掉两侧挡板,将凝胶放入盛有电泳液的槽中(加样孔朝向负极端,DNA由负极向正极移动),使液面高出凝胶2~3mm,小心拔出梳子。 5.DNA样品与载体缓冲液5:1混合并加入凹孔中(样品不可溢出)。 6.打开电源,调节所需电压,电压与凝胶的长度有关,一般使用电压不超过5v/cm。 7.据指示染料移动的位置,确定电泳是否终止(溴酚蓝的涌动距离在5sRNA和0.3kbDNA 带之间)。 8.电泳完毕关闭电源。将凝胶放紫外灯下观察并拍照。 三、目的DNA片段基因扩增(PCR) 方案1:遗传学实验31,PCR扩增基因片段 方案2: 【实验目的】 了解多聚合酶链反应DNA 扩增技术的基本原理和实验应用,掌握PCR反应基本技术。【实验原理】 PCR(Polymerase Chain Reaction)即聚合酶链式反应是1986 年由Kallis Mullis 发现。这项技术已广泛地应用于分子生物学各个领域,它不仅可用于基因分离克隆和核酸序列分析,还可用于突变体和重组体的构建,基因表达调控的研究,基因多态性的分析,遗传病和传染病诊断,肿瘤机制探查,法医鉴定等方面。PCR技术已成为方法学上的一次革命,它必将
基因克隆的几种常见方法
基因克隆的几种常见方法 基因(gene)是遗传物质的最基本单位,也是所有生命活动的基础。不论要揭示某个基因的功能,还是要改变某个基因的功能,都必须首先将所要研究的基因克隆出来。特定基因的克隆是整个基因工程或分子生物学的起点。本文就基因克隆的几种常用方法介绍如下。 1 根据已知序列克隆基因 对已知序列的基因克隆是基因克隆方法中最为简便的一种。获取基因序列多从文献中查取,即将别人报道的基因序列直接作为自己克隆的依据。现在国际上公开发行的杂志一般都不登载整个基因序列,而要求作者在投稿之前将文章中所涉及的基因序列在基因库中注册,拟发表的文章中仅提供该基因在基因库中的注册号(accession number),以便别人参考和查询。目前,世界上主要的基因库有1)EMBL,为设在欧洲分子生物学实验室的基因库,其网上地址为 https://www.360docs.net/doc/5d16308285.html,/ebi-home.html;(2)Genbank,为设在美国国家卫生研究院(NIH)的基因库,其网上地址为 https://www.360docs.net/doc/5d16308285.html,/web/search/index.html;(3)Swissport和TREMBL,Swissport是一蛋白质序列库,其所含序列的准确度比较高,而TREMBL只含有从EMBL库中翻译过来的序列。目前,以Genbank的应用最频繁。这些基因库是相互联系的,在Genbank注册的基因序列,也可能在Swissport注册。要克隆某个基因可首先通过Internet查询一下该基因或相关基因是否已经在基因库中注存。查询所有基因文库都是免费的,因而极易将所感兴趣的基因从库中拿出来,根据整个基因序列设计特异的引物,通过PCR从基因组中克隆该基因,也可以通过RT-PCR克隆cDNA。值得注意的是,由于物种和分离株之间的差异,为了保证PCR 扩增的准确性,有必要采用两步扩增法,即nested PCR。 根据蛋白质序列也可以将编码该蛋白质的基因扩增出来。在基因文库中注册的蛋白质序列都可以找到相应的DNA或cDNA序列。如蛋白质序列是自己测定的,那么需要设计至少1对简并引物(degenerated primer),从cDNA文库中克隆该基因。以这种方法克隆的基因必须做序列测定才能鉴别所扩增产物的特异性。 另外,在基因克隆之后,如还要进一步做表达研究,所使用的PCR酶最好不用Taq DNA聚合酶,而采用其他有自我检测(reading proof)功能的酶,如pfu。这样可以避免由于扩增过程中出现的点突变或终止密码子而导致整个研究结论的错误。 2 根据已知探针克隆基因 这也是基因克隆的一种较直接的方法。首先将探针作放射性或非放射性标记,再将其与用不同内切酶处理的基因组DNA杂交,最后将所识别的片段从胶中切下来,克隆到特定的载体(质粒、噬菌体或病毒)中作序列测定或功能分析。这种方法不但可以将基因克隆出来,还能同时观察该基因在基因组中的拷贝数。但在探
疾病基因克隆的策略及主要方法
疾病基因克隆的策略及主要方法 申海鹰周元国(第三军医大学野战外科研究所分子生物学中心,重庆400042) 摘要疾病基因的分离和克隆是功能基因组学的研究热点,具体策略的选择取决于疾病背景资料的掌握程度,为能快速、准确地克隆出目的基因,本文介绍两类常用的基因克隆策略——定位克隆策略、功能克隆策略——及其主要方法,如:家系连锁分析、等位基因共占法、人群相关分析法、抑制性消减杂交、差示反转录PCR、差异消减显示法、代表性差异分析法、比较基因组杂交等,并作简要的评价。 关键词基因;定位克隆;消减杂交 学科分类号Q785 Strategies and methods for cloning pathogenic gene SHEN Hai-ying, ZHOU Yuan-guo. (C enter of Molecular Biology, Research Institute of Surgery and Daping Hospital, Third Milit ary Medical University, Chongqing 400042) Abstract Isolation and cloning of pathogenic gene is a hot spot in functional genome stu dy, while it is the disease background which decides the selection of the strategies. Tw o strategies, mapping strategy and functional cloning strategy, which can clone the obje ctive gene rapidly and accurately were introduced. Some main methods including family-based linkage analysis, allele sharing method, population association analysis, suppressio n subtractive hybridization (SSH), differential display reverse-transcription PCR (DD-RT-PC R), differential subtraction display (DSD), representational difference analysis (RDA), com parative genome hybridization (CEH) were elucidated briefly. Key words: Gene; Mapping clone; Subtractive hybridization 基因组全序列测定可望提前完成,而以功能鉴定为中心的功能基因组学应运而生,将人类5~10万个基因定位及克隆是一项庞大而艰巨的任务。自1911年Wilson将色盲基因定位于X染色体起,随着连锁分析方法的发展和体细胞杂交、重组DNA、分子杂交以及PCR技术的发现和应用,陆续出现了几种改进或全新的遗传学基因定位和克隆方法。与此同时,另一类以消减杂交为基本原理的代表性差异分析、基因组错配扫描、比较基因组杂交及mRNA差示等方法的出现和应用,使一些多基因遗传病相关致病基因的筛查和定位面临突破。迄今为止,约有5000个遗传性状被定位,其中400多个为致病基因[1]。根据不同的背景资料,人类基因克隆可采取的思路有以下四种(见附图): 目前人类基因克隆的主要策略有三种:一是反向遗传学定位克隆策略,它通过RFLP、微卫星D NA等遗传标记,先获得某一表型基因在染色体上的定位,再在候选区域内选择已知基因,进行致病突变的筛选,并获得cDNA及全基因;另一类是从蛋白质功能着手的功能克隆策略,采用以消减杂交为策略的多种分子生物学手段,先通过消减获得特异表达或缺失的基因片段,然后进行染色体定位乃至获得全基因。本文拟就前两种主要策略和各自方法的优缺点作一介绍和分析。此外,尚有介于两者之间的候选克隆策略,包括定位候选克隆和功能候选克隆,前者是在将疾病基因以连锁分析和染色体分析基本定位以后,再在候选区域内选择所有已知基因进行致病突变的筛