寻找差异表达的基因

基因表达谱数据
基因表达谱可以用一个矩阵来表示,每一行代表一个基因,每一列代表一个样本(如图1)。所有基因的表达谱数据在“gene_exp.txt ”文件中存储,第一列为基因的entrez geneid ,第2~61列是疾病样本的表达,第62~76列是正常样本的表达。
图1 基因表达谱的矩阵表示
寻找差异表达的基因:
原理介绍:
差异表达分析是目前比较常用的识别疾病相关miRNA 以及基因的方法,目前也有很多差异表达分析的方法,但比较简单也比较常用的是Fold change 方法。它的优点是计算简单直观,缺点是没有考虑到差异表达的统计显著性;通常以2倍差异为阈值,判断基因是否差异表达。Fold change 的计算公式如下:
normal
Disease x x c Fold =
_
即用疾病样本的表达均值除以正常样本的表达均值。
差异表达分析的目的:识别两个条件下表达差异显著的基因,即一个基因在两个条件中的表达水平,在排除各种偏差后,其差异具有统计学意义。我们利用一种比较常见的T 检验(T-test )方法来寻找差异表达的miRNA 。T 检验的主要原理为:对每一个miRNA 计算一个T 统计量来衡量疾病与正常情况下miRNA 表达的差异,然后根据t 分布计算显著性p 值来衡量这种差异的显著性,T 统计量计算公式如下:
n
s n s x x t normal Disease normal
Disease miRNA //22+-=
对于得到的显著性p 值,我们需要进行多重检验校正(FDR ),比较常用的是BH 方法(Benjamini and Hochberg, 1995)。
1+
=N
v
t分布
程序实现:
●基因表达谱数据--- gene_exp.txt
●Matlab软件实现mRNA差异表达分析:
MATLAB软件安装好之后,双击系统桌面的MATLAB图标,或在开始菜单的程序选项中选择MATLAB快捷方式,即开始启动MATLAB。初次启动MATLAB后,将进入MATLAB默认设置的桌面平台。桌面平台包括命令窗口、历史窗口、当前目录窗口和工作间管理窗口等窗口(如图2)。
图2 matlab窗口简介
工作空间主要包含了目前用户定义的一些变量,用户可以在命令窗口执行一些特定的命令操作来完成特定的功能。我们首先将工作目录选择到我们数据存放的硬盘目录下,然后导入要分析的基因表达谱数据,进行差异表达分析。
在命令窗口输入main_MTDN_end.m程序中的1-21行命令(注意要将程序中的目录改变到自己数据的存储目录下),即可得到差异表达的基因。这段程序主要包含两个函数:mattest和mafdr。
mattest函数是进行t检验的,输入的数据为疾病和正常的表达谱数据,返回每个miRNA的T统计量和对应的p值。这个参数还可以利用…Permute?参数进行随机扰动,'Showhist'参数用来显示T统计量和p值的分布。
mafdr函数是用来计算FDR的函数,可以利用参数来选择计算FDR的方法,这里我们利用“BHFDR”参数来选择BH方法对p值进行校正,利用'showplot'参数来显示FDR的图示结果。
结果可以在工作空间窗口中通过双击变量进行查看。
结果展示:
T-统计量和p值的分布图以及FDR:
图3 T-score,P-values以及FDR的分布
●差异表达mRNA:我们卡的阈值为FDR<0.1;2倍fold change
(Fold_c>2 or <1/2 ),我们识别了11个下调的mRNA和6个上调的mRNA。
差异表达基因的层次聚类分析
mRNA表达谱数据:差异表达17个mRNA的表达数据
程序实现:
我们接下来利用差异表达mRNA的表达谱进行聚类分析,在命令窗口输入main_MTDN_end.m程序中的23-30行命令,结果会输出利用差异表达mRNA聚类分析的结果。这部分主要是利用一个现有的函数clustergram进行聚类分析,函数的输入数据是差异表达mRNA的表达谱。之后可以利用set 函数对行的符号和列的符号进行设定。
聚类分析结果展示:
聚类做heatmap,我比较喜欢用pheamap,简单又好看,但是很多做heatmap
的函数都不带输出聚类后基因名字的功能。heatmap旁标注基因是很有用的信息,
论文中经常会用到,所以我们可以更改pheatmap的源代码,让它输出基因列表,
其实如果能够给出基因list,在heatmap旁边标注出list中的基因就好了,但有了基因列表也可以做这个事情。
从cran上下载pheatmap的源代码,打开pheatmap的R文件夹中pheatmap.R文件,在一大串#上面添加write_matrix = function( mat,
out_file ){
write.table(as.data.frame(mat),sep="\t",quote=FALSE, file=out_file)
}
在一大串#下面的pheatmap中添加out_file = NA,此乃默认参数设定。
在hclust之后,就是当cluster_mat函数处理了mat矩阵后,添加
if( !is.na(out_file) ){
write_matrix( mat, out_file )
}
**************************************************昏割线
********************************************************
打开Rstudio,tools--install packages--选择那个压缩包ok啦
用法:
>setwd("F:/project/PTEN/01.RPKM/correlation")
>dataframe <- read.table("correlation.txt",header=TRUE)
>pheatmap(dataframe,color=colorRampPalette(c("steelblue1","black","yel low"))(50),out_file="F:/project/gene/new.txt")
基因名字输出到new.txt中了
基因差异表达的研究方法
基因差异表达的研究方法 摘要寻找差异表达基因成为目前基因研究的一个非常重要的手段。寻找差异表达基因的方法有消减杂交法、mRNA 差异显示、代表性差异分析法、基因表达的序列分析、抑制消减杂交、表达序列标签、cDNA微阵列、半定量PCR、定量PCR。特综述以上各种方法的原理、方法过程、优缺点及其应用,随着科学技术的发展对差异表达基因的研究会更加完善。 关键词基因;差异表达;消减杂交;差异显示;研究方法 在真核生物的生命现象中,从个体的发育、生长、衰老、死亡,到组织、细胞的分化、凋亡或肿瘤的恶化以及细胞对各种生物、理化因子的应答,本质上都涉及基因在时间上或空间上的选择性表达,即基因的差异表达。基因的差异表达与组织、细胞的生物学性状和功能密切相关,成为生命科学的重要研究课题(潘美辉等,1997)。比较不同细胞或不同基因型在基因表达上的差异,不仅是研究生命过程分子机制的基础,亦是分离克隆目的基因的前提(胡昌华,2001)。寻找差异表达基因成为目前基因研究的一个非常重要的内容。差异表达的基因通常用稳定状态下mRNA的丰度高低有无来比较。差异表达基因有2个含义,即表达基因的种类改变和基因表达量的变化。通过它能找到疾病不同阶段、不同状态下表达不同丰度的基因,从而为进一步研究打下基础。分离和鉴定差异表达基因是了解各项生命活动和疾病分子调控机制的重要手段(梁自文,2001)。笔者拟对目前现有的寻找差异基因的方法作一综述。 1消减杂交法(subtractive hybridization) 消减杂交在1984年由Palmer和Lamer(Lamar EE et at.,1984)提出,其目的是分离出两类同源分子间差异表达的基因,关键是利用分子杂交原理去除共同序列,保留差异序列,通过PCR多次循环扩增而分离,从而能进一步研究其差异表达基因。 具体做法:首先以oligo-dT为引物,从tester中制备放射性标记的单链cDNA 文库。然后将这些cDNA探针与过量的来自driver的mRNA(其poly-A尾已与生物素耦联)杂交,大部分单链cDNA探针和driver中的mRNA形成异源双链,并通过羟基磷灰石柱层除去cDNA×mRNA杂交体,以此富增tester中特异的cDNA。消减杂交法的最大优点是它适用于未被克隆的基因组片段;其次它特别适于寻找那些由于缺失造成突变的基因。但这一方法需要大量的driver mRNA才能使消减杂交充分进行,所回收的cDNA量也很低,而且操作步骤复杂、耗资
三种差异表达基因克隆方法的比较
三种差异表达基因克隆方法的比较 [摘要] 为了能快速、有效、准确地克隆出有意义的基因,本文就常用的三种基因克隆方法,即差异显示PCR、抑制性消减杂交、PAP-PCR,从其原理、应用、优越性、主要缺陷等几方面进行了简要概述。这些方法为中医、针刺治疗各种疾病过程中有关的基因差异表达,以及有关基因分子克隆提供有效的分子生物学工具。 [主题词] 针灸原理;基因表达;克隆,分子;聚合酶链反应 高等生物大约有100000个不同的基因,但在生物体内任一细胞中只有15%的基因得以表达。而这大约15000个基因的表达是按时间和空间顺序有序地进行着,这种表达方式即为基因的差别表达(differentialexpression)[1]。生物体表现出各种各样的特性,主要是其基因表达的差异引起的。基因差异表达的变化有两种,即新出现的基因表达与表达量差异的基因表达。由于基因表达的变化是调控细胞生命活动过程的核心机制,通过比较同一类细胞在不同生理状态下或在不同生长发育阶段的基因表达差异,可为分析生命活动过程提供重要信息。当前,人类基因组研究的重心正在由“结构”向“功能”转移,一个以基因组功能研究为主要内容的所谓 “后基因组时代”(post genomics),即功能基因组(func tionalgenomics)时代即将到来,这就决定了寻找差异表达基因成为分子生物学的研究热点之一。 目前已有针灸科研工作者从分子生物学水平探讨针灸治疗各种疾病的可能机理。有实验[2]报道针刺翳风等穴位面神经组织中NT 3及TrKCmR NA表达增加;针刺后癫痫大鼠海马内nNOSmRNA和iNOSmRNA增加[3];针刺可调节CCK 8mRNA、THmRNA、SOMmRNA、c fos\c junmRNA、CGRPmRNA的 增减[4]。为适应当前医学的发展,为满足针灸科研工作者分子生物学研究的需要,本文对差异显示PCR、抑制性消减杂交及RNA任意引物PCR三种常用的差异基因筛选法,分别从其基本原理、过程、应用范围及优缺点等作一个简介。 1 差异显示PCR(differentialdisplayreverse tran scriptasePCR,DDRT PCR) 该方法是自1992年由Liang和Pardee建立起来的[1],后经多年改进,目前已被广泛的使用。它已较成功地用于胚胎发生[5]、脑发育[6]、生长因子激活与抑
基因表达的检测的几种方法
基因表达检测的最终技术目标是能确定所关注的任何组织、细胞的 RNA的绝对表达量。可以先从样本中抽提RNA,再标记RNA, 然后将这些标记物作探针与芯片杂交,就可得出原始样本中不同 RNA的量。然而用于杂交的某个特定基因的RNA的量与在一个 相应杂交反应中的信号强度之间的关系十分复杂,它取决于多种 因素,包括标记方法、杂交条件、目的基因的特征和序列。所以 芯片的方法最好用于检验两个或多个样本中的某种RNA的相对 表达量。样本之间某个基因表达的差异性(包括表达的时间、空 间特性及受干扰时的改变)是基因表达最重要的,而了解RNA 的绝对表达丰度只为进一步的应用或多或少地起一些作用。 基因表达的检测有几种方法。经典的方法(仍然重要)是根据在 细胞或生物体中所观察到的生物化学或表型的变化来决定某一 特定基因是否表达。随着大分子分离技术的进步使得特异的基因 产物或蛋白分子的识别和分离成为可能。随着重组DNA技术的 运用,现在有可能检测.分析任何基因的转录产物。目前有好几 种方法广泛应用于于研究特定RNA分子。这些方法包括原位杂交.NORTHERN凝胶分析.打点或印迹打点.S-1核酸酶分 析和RNA酶保护研究。这里描述RT-PCR从RNA水平上检查 基因表达的应用。8 f3 f- |2 L) K) b7 ]- ~- | RT-PCR检测基因表达的问题讨论
关于RT-PCR技术方法的描述参见PCR技术应用进展,在此主要讨论它在应用中的问题。理论上1μL细胞质总RNA对稀有mRNA扩增是足够了(每个细胞有1个或几个拷贝)。1μL差不多相当于50-100,000个典型哺乳动物细胞的细胞质中所含RNA的数量,靶分子的数量通常大于50,000,因此扩增是很容易的。该方法所能检测的最低靶分子的数量可能与通常的DNAPCR相同;例如它能检测出单个RNA分子。当已知量的转录RNA(用T7RNA聚合酶体外合成)经一系列稀释,实验结果表明通过PCR的方法可检测出10个分子或低于10个分子,这是反映其灵敏度的一个实例。用此技术现已从不到1个philadelphia染色体阳性细胞株K562中检测到了白血病特异的MRNA的转录子。因此没必要分离polyA+RNA,RNA/PCR法有足够的灵敏度来满足绝大多数实验条件的需要。 7 H+ F& _* S6 W( a8 p: [, @- d, { 将PCR缓冲液同时用于反转录酶反应和PCR反应,可简化实验步骤。我们发现整个反应过程皆用PCR缓冲液的结果相当于或优于先用反转录缓冲液合成CDNA,然后PCR缓冲液进行PCR扩增循环。当然,值得注意的是PCR缓冲液并不最适合第一条DNA链的合成。我们对不同的缓冲液用于大片段DNA 合成是否成功还没有进行过严格的研究。
基因差异表达技术
基因差异表达技术 真核生物中,从个体的生长、发育、衰老、死亡,到组织的得化、调亡以及细胞对各种生物、理化因子的应答,本质上都涉及基因的选择性表达。高等生物大约有30000个不同的基因,但在生物体内任意8细胞中只有10%的基因的以表达,而这些基因的表达按特定的时间和空间顺序有序地进行着,这种表达的方式即为基因的差异表达。其包括新出现的基因的表达与表达量有差异的基因的表达。生物体表现出的各种特性,主要是由于基因的差异表达引起的。 由于基因的差异表达的变化是调控细胞生命活动过程的核心机制,通过比较同一类细胞在不同生理条件下或在不同生长发育阶段的基因表达差异,可为分析生命活动过程提供重要信息。研究基因差异表达的主要技术有差别杂交(differential hybridization)、扣除(消减)杂交(subtractive hybridization of cDNA,SHD)、mRNA差异显示(mRNA differential display,DD)、抑制消减杂交法(suppression subtractive hybridization,SSH)、代表性差异分析(represential display analysis,RDA)、交互扣除RNA差别显示技术(reciprocal subtraction differential RNA display)、基因表达系列分析(serial analysis of gene expression,SAGE)、电子消减(electronic subtraction)和DNA微列阵分析(DNA microarray)等。 一、差别杂交与扣除杂交 差别杂交(differential hybridization)又叫差别筛选(differential screening),适用于分离经特殊处理而被诱发表达的mRNA的cDNA克隆。为了增加这种方法的有效性,后来又发展出了扣除杂交(subtractive hybridization)或扣除cDNA克隆(subtractive cDNA cloning),它是通过构建扣除文库(subtractive library)得以实现的。 (一)差别杂交 从本质上讲,差别杂交也是属于核酸杂交的范畴。它特别适用于分离在特定组织中表达
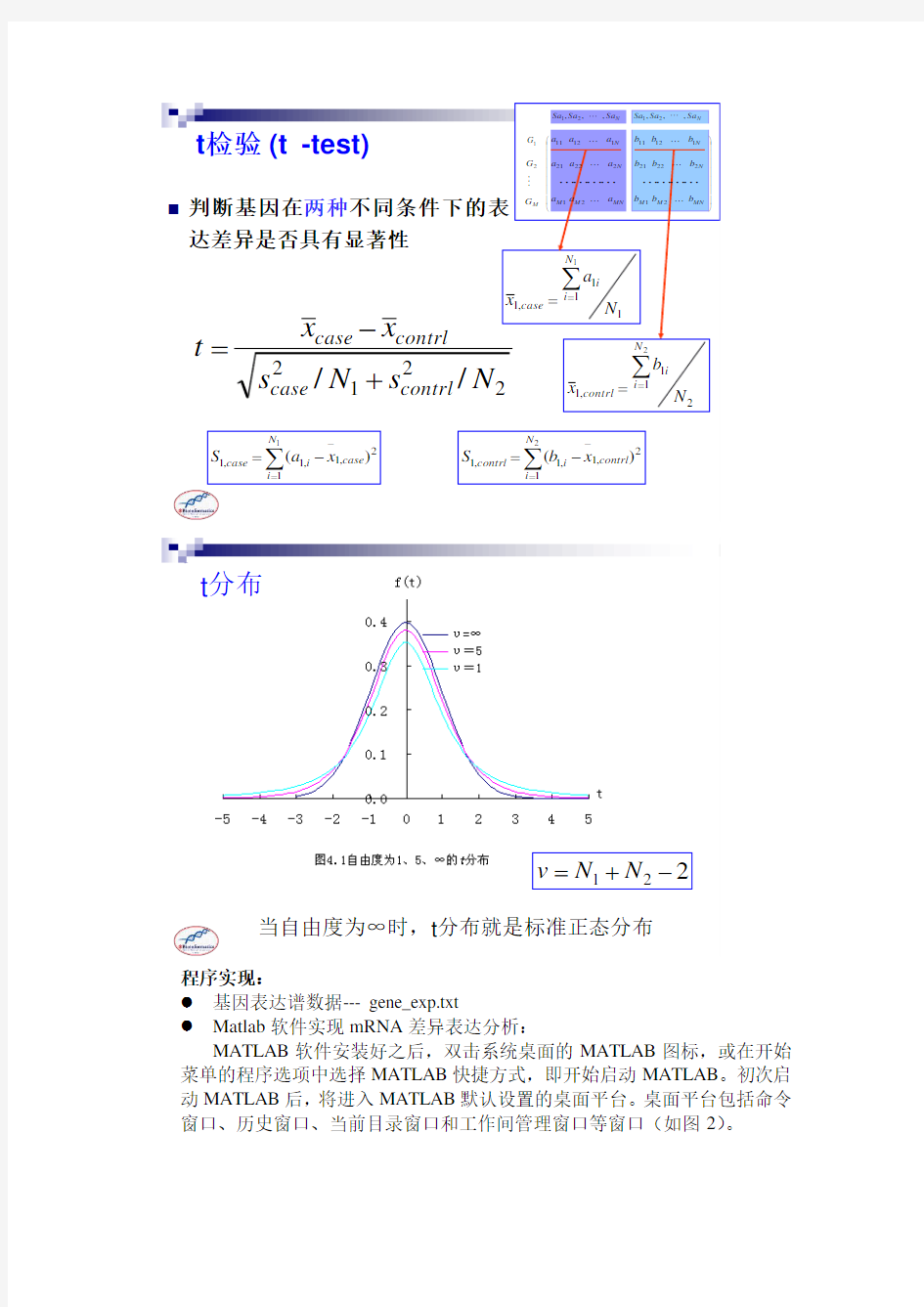
寻找差异表达的基因
基因表达谱数据 基因表达谱可以用一个矩阵来表示,每一行代表一个基因,每一列代表一个样本(如图1)。所有基因的表达谱数据在“gene_exp.txt ”文件中存储,第一列为基因的entrez geneid ,第2~61列是疾病样本的表达,第62~76列是正常样本的表达。 图1 基因表达谱的矩阵表示 寻找差异表达的基因: 原理介绍: 差异表达分析是目前比较常用的识别疾病相关miRNA 以及基因的方法,目前也有很多差异表达分析的方法,但比较简单也比较常用的是Fold change 方法。它的优点是计算简单直观,缺点是没有考虑到差异表达的统计显著性;通常以2倍差异为阈值,判断基因是否差异表达。Fold change 的计算公式如下: normal Disease x x c Fold = _ 即用疾病样本的表达均值除以正常样本的表达均值。 差异表达分析的目的:识别两个条件下表达差异显著的基因,即一个基因在两个条件中的表达水平,在排除各种偏差后,其差异具有统计学意义。我们利用一种比较常见的T 检验(T-test )方法来寻找差异表达的miRNA 。T 检验的主要原理为:对每一个miRNA 计算一个T 统计量来衡量疾病与正常情况下miRNA 表达的差异,然后根据t 分布计算显著性p 值来衡量这种差异的显著性,T 统计量计算公式如下: n s n s x x t normal Disease normal Disease miRNA //22+-= 对于得到的显著性p 值,我们需要进行多重检验校正(FDR ),比较常用的是BH 方法(Benjamini and Hochberg, 1995)。
基因表达差异分析方法进展
高等真核生物的基因组一般具有80 000~100 000个基因,而每一个细胞大约只表达其中的15%[1]。基因在不同细胞间及不同生长阶段的选择性表达决定了生命活动的多样性,如发育与分化、衰老与死亡、内环境稳定、细胞周期调控等。比较细胞间基因表达的差异为我们揭示生命活动的规律提供了依据。 由于真核细胞mRNA 3′端一般含有Poly(A)尾,因此现有的方法基本上都是利用共同引物将不同的mRNA反转录成cDNA,以cDNA为对象研究基因表达的差异。1992年Liang等[2]建立了一种差异显示反转录PCR法(differential display reverse transcription PCR,DDRT-PCR),为检测成批基因表达的差异开辟了新天地。迄今为止已出现了大量应用该技术的研究报道[3,4]。然而,尽管应用DDRT-PCR方法已经取得了不少成果,而且该方法还在不断改进之中,但它仍然存在几个难以解决的问题:(1) 重复率低,至少有20%的差异条带不能被准确重复[5];(2) 假阳性率可以高达90%[6];(3) 获得的差异表达序列极少包含编码信息。近年来,针对DDRT-PCR方法的不足,又有几种新的检测差异表达基因的方法出现,现仅就这方面的进展做一简要介绍。 1.基因表达指纹(gene expression fingerprinting,GEF):GEF技术使用生物素标记的引物Bio-T13合成cDNA第一链,用dGTP对其进行末端加尾,再以富含C的引物引发合成cDNA第二链。用限制性内切酶消化双链cDNA,以交联有抗生物素蛋白的微球捕获cDNA3′端,以T4DNA连接酶连接同前述内切酶相对应的适配子,并以Bio-T13及适配子中的序列作为新的引物进行特异的PCR 扩增,得到大量的特异cDNA片段。适配子末端被32P-dATP标记后,固定于微球上的cDNA片段经过一系列酶切,产生的酶切片段从微球表面释放出来,其中那些含有标记末端的片段经凝胶电泳后构成mRNA指纹图谱。通过分析不同细胞间的指纹图谱就能得到差异表达的序列[7]。GEF技术所需的工作量较DDRT-PCR明显减少,由于用酶切反应替代了条件不严格的PCR反应,其重复性也较好,假阳性率低,并且所获得的片段中包含有一定的编码信息。GEF技术最大的缺点在于电泳技术的局限。由于它的指纹图谱要显示在同一块电泳胶上,经过几轮酶切之后常会得到1 000~2 000条电泳带,而现有的PAGE电泳很少能分辨超过400条带,故只有15%~30%的mRNA能够被辨认出来,因此得
基因表达及分析技术
基因表达及其分析技术 生命现象的奥秘隐藏在基因组中,对基因组的解码一直是现代生命科学的主流。基因组学研究可以说是当今生命科学领域炙手可热的方向。从DNA 测序到SNP、拷贝数变异(copy number variation , CNV)等DNA多态性分析,到DNA 甲基化修饰等表观遗传学研究,生命过程的遗传基础不断被解读。 基因组研究的重要性自然不言而喻。应该说,DNA 测序技术在基因组研究 中功不可没,从San ger测序技术到目前盛行的新一代测序技术(Next Gen eration Seque ncing NGS)到即将走到前台的单分子测序技术,测序技术是基因组解读最重要的主流技术。而基因组测序、基因组多态性分析、DNA 甲基化修饰等表观遗传分析等在基因组研究中是最前沿的课题。但是基因组研究终究类似“基因算命”,再清晰的序列信息也无法真正说明一个基因的功能,基因功能的最后鉴定还得依赖转录组学和蛋白组学,而转录作为基因发挥功能的第一步,对基因功能解读就变得至关重要。声称特定基因、特定SNP、特定CNV、特定DNA修饰等与某种表型有关,最终需要转基因、基因敲除、突变、 RNAi 、中和抗体等技术验证,并必不可少要结合基因转录、翻译和蛋白修饰等数据。 基因实现功能的第一步就是转录为mRNA或非编码RNA,转录组学主要研究基因转录为RNA 的过程。在转录研究中,下面几点是必须考虑的: 1,基因是否转录(基因是否表达)及基因表达水平高低(基因是低丰度表达还是中、高丰度表达)。特定基因有时候在一个细胞中只有一个拷贝的表达,而表达量会随细胞类型不同或发育、生长阶段不同或生理、病理状态不同而改变。因此任何基
基因表达分析
荧光定量PCR 在基因表达分析中的应用 所谓基因表达就是指在特定的时刻某种我们感兴趣的基因在组织或细胞中的mRNA 的表达数量。众所周知,很多的疾病(如肿瘤)的发生发展、很多药物的作用机理、很多生物的代谢调控作用等都和基因表达的变化有关,因此对基因表达进行精确定量是十分重要的。过去为了对mRNA 进行定量有了各种各样的方法,如Southern 杂交、Northern 杂交、原位杂交、传统PCR 等,但是我们也都知道这些技术灵敏性较差,重复性不好,操作比较烦琐,已经无法满足现在科研和检测的需要,于是荧光定量PCR 技术也就应运而生了。荧光定量PCR 技术能对核酸进行精确定量,因此大大提高了在基因表达的准确性和灵敏度,深受用户的青睐,广泛的应用于肿瘤研究、药物筛选、功能基因组研究等各个领域,目前已经成了很多科研文章发表的重要实验内容。 基因表达分析中常见到的重要问题 1、要检测的基因 基因表达分析的目的就是检测某种我们感兴趣的基因在不同组织或细胞中的表达差异。荧光定量PCR 技术可以对核酸物质的含量进行精确的定量,也就成了研究基因表达差异的一把利器。 在基因表达分析实验中要检测两个基因,一个是目的基因和另一个是看家基因。之所以要引入看家基因是由于不能确定要比较的样品所用的组织起始量相同。就是说比如有的老师提取正常样品的基因时用了100个细胞,而提取病变样品时只用了10个细胞,这时候的基因表达差异可能是由于提取时候的样品细胞数不同引起的,为了纠正这种误差,我们选用认为在两个样本中表达量不变的基因作为内参照,来去除这带来的干扰。例如,要研究某个基因在肿瘤样品和正常样品中的基因表达差异。我们在实验中发现我们选择研究的正常样品中的看家基因的表达量是肿瘤样品中的10倍,就认为正常样品的细胞数就是肿瘤样品细胞数的10倍,那么在肿瘤样品中目的基因的基因表达量应该乘以10倍,才能和正常样品进行比较。 2、计算基因表达差异 基因表达差异的计算是通过所得到的Ct 值来计算的,要计算两个样品(待测样品和对照样品)的目的基因的表达差异必须检测得到4个Ct 值:待测样品和对照样品中目的基因和看家基因的Ct 值。 那么基因表达差异应该计算为 基因表达差异=2(△Ct1-△Ct2) 目的基因 看家基因 待测样品 对照样品 △Ct1 △Ct2
【R高级教程】专题二:差异表达基因的分析
【R高级教程】专题二:差异表达基因的分析 应学生及个别博友的要求,尽管专业博文点击率和反应均很差,但在去San Diego参加PAG会议之前,还是抽时间给出【R高级教程】的第二专题。专题一给出了聚类分析的示例,本专题主要谈在表达谱芯片分析中如何利用Bioconductor鉴定差异表达基因。 鉴定差异表达基因是表达谱芯片分析pipeline中必须的分析步骤。差异表达基因分析是根据表型协变量(分类变量)鉴定组间差异表达,它属于监督性分类的一种。在鉴定差异表达基因以前,一般需要对表达值实施非特异性过滤(在机器学习框架下属于非监督性分类),因为适当的非特异性过滤可以提高差异表达基因的检出率、甚至是功效。R分析差异表达基因的library有很多,但目前运用最广泛的Bioconductor包是limma。 本专题示例依然来自GEO数据库中检索号为GSE11787 的Affymetrix芯片的数据,数据介绍参阅专题一。 >library(limma) >design <- model.matrix(~ -1+factor(c(1,1,1, 2,2,2))) 这个是根据芯片试验设计,对表型协变量的水平进行design,比如本例中共有6张芯片,前3张为control对照组,后3张芯片为实验处理组,用1表示对照组,用2表示处理组。其他试验设计同理,比如2*2的因子设计试验,如果每个水平技术重复3次,那么可以表示为:design <- model.matrix(~ -1+factor(c(1,1,1, 2,2,2, 3,3,3, 4,4,4)))。接上面的程序语句继续:
>colnames(design) <- c("control", "LPS") >fit <- lmFit(eset2, design) >contrast.matrix <- makeContrasts(control-LPS, levels=design) >fit <- eBayes(fit) >fit2 <- contrasts.fit(fit, contrast.matrix) >fit2 <- eBayes(fit2) >results<-decideTests(fit2, method="global", adjust.method="BH", p.value=0.01, lfc=1.5) >summary(results) >vennCounts(results) >vennDiagram(results) 比较遗憾的是,目前limma自带的venn作图函数不能做超过3维的高维venn图,只能画出3个圆圈的venn图,即只能同时对三个
基因表达谱分析技术
基因表达谱分析技术 1、微阵列技术(microarray) 这是近年来发展起来的可用于大规模快速检测基因差别表达、基因组表达谱、DNA序列多态性、致病基因或疾病相关基因的一项新的基因功能研究技术。其原理基本是利用光导化学合成、照相平板印刷以及固相表面化学合成等技术,在固相表面合成成千上万个寡核苷酸“探针”(cDNA、ESTs或基因特异的寡核苷酸),并与放射性同位素或荧光物标记的来自不同细胞、组织或整个器官的DNA或mRNA反转录生成的第一链cDNA进行杂交,然后用特殊的检测系统对每个杂交点进行定量分析。其优点是可以同时对大量基因,甚至整个基因组的基因表达进行对比分析。包括cDNA芯片(cDNA microarray)和DNA 芯片(DNA chips)。 cDNA芯片使用的载体可以是尼龙膜,也可以是玻片。当使用尼龙膜时,目前的技术水平可以将20000份材料点在一张12cm×18cm的膜上。尼龙膜上所点的一般是编好顺序的变性了的双链cDNA片段。要得到基因表达情况的数据,只需要将未知的样品与其杂交即可。杂交的结果表示这一样品中基因的表达模式,而比较两份不同样品的杂交结果就可以得到在不同样品中表达模式存在差异的基因。杂交使用的探针一般为mRNA的反转录产物,标记探针使用32PdATP。如果使用玻片为载体,点阵的密度要高于尼龙膜。杂交时使用两种不同颜色的荧光标记不同的两份样品,然后将两份样品混合起来与一张芯片杂
交。洗去未杂交的探针以后,能够结合标记cDNA的点受到激发后会发出荧光。通过扫描装置可以检测各个点发出荧光的强度。对每一个点而言,所发出的两种不同荧光的强度的比值,就代表它在不同样品中的丰度。一般来讲,显示出来的图像中,黄色的点表示在不同的样品中丰度的差异不大,红色和绿色的点代表在不同样品中其丰度各不相同。使用尼龙膜为载体制作cDNA芯片进行研究的费用要比玻片低,因为尼龙膜可以重复杂交。检测两种不同的组织或相同组织在不同条件下基因表达的差异,只需要使用少量的尼龙膜。但是利用玻片制作的cDNA芯片灵敏度更高,而且可以使用2种探针同时与芯片杂交,从而降低了因为杂交操作带来的差异;缺点是无法重复使用还必须使用更为复杂的仪器。 Guo等(2004)将包含104个重组子的cDNA文库点在芯片上,用于检测拟南芥叶片衰老时的基因表达模式,得到大约6200差异表达的ESTs,对应2491个非重复基因。其中有134个基因编码转录因子,182个基因预测参与信号传导,如MAPK级联传导路径。Li等(2006)设计高密度的寡核苷酸tiling microarray方法,检测籼稻全基因组转录表达情况。芯片上包含13,078,888个36-mer寡核苷酸探针,基于籼稻全基因组shot-gun测序的序列合成,大约81.9%(35,970)的基因发生转录事件。Hu等(2006)用含有60,000寡核苷酸探针(代表水稻全部预测表达基因)的芯片检测抗旱转基因植株(过量表达SNAC1水稻)中基因的表达情况,揭示大量的逆境相关基因都是上升表达的。 2、基因表达系列分析(Serial analysis of gene expression, SAGE)
检测差异表达基因技术的研究进展
[收稿日期]1999204219;[修回日期]1999207227 [作者简介]金慧英(1966-)女,江苏南京市人,助理研究员,硕士,主要从事微生物学方面的研究。 [文章编号]100028861(2000)04(S )20111203 检测差异表达基因技术的研究进展 Advance of study on the iden tify i ng d ifferen ti a l expressi ng gene 金慧英(J I N H u i 2ying ),房德兴(FAN G D e 2x ing ) (南京军区军事医学研究所,江苏南京210002) [摘 要] 随着差异表达基因检测技术的不断发展,建立了很多新的检测方法,本文对mRNA 差异显示技术(DDR T 2 PCR )、c DNA 示差分析技术(c DNA 2RDA )及抑制消减杂交技术(SSH )的基本原理及工作流程作了综述,并介绍了它们的应用状况和特点,这些方法的建立和广泛应用可望发现更多的差异表达的基因,并通过其与表型的关系分析加快人们对基因功能的认识。 [关键词] mRNA 差异显示(DDR T 2PCR );c DNA 示差分析(c DNA 2RDA );抑制消减杂交(SSH )[中图分类号] Q 78 [文献标识码] A 近年来现代分子生物学技术,特别是核酸杂交技术、反转录技术、PCR 扩增技术的发展为建立简便高效的检测差异表达基因的方法奠定了坚实的基 础。从L I AN G (1992)[1] 建立的m RNA 差异显示技术(DDR T 2PCR ),L IS IT SYN (1993)[2]建立并由 HU BAN K (1994)[3] 改进的c DNA 示差分析技术 (c DNA 2RDA )到D I A TCH EN KU (1996)[4] 建立的抑制消减杂交法(SSH ),差异表达基因的检测技术越来越简便,且特异性更强,灵敏度更高。现就上述3种方法的原理、工作流程、应用及展望简述如下:1 基本原理及工作流程 111 m RNA 差异显示技术(m RNA differen tial disp lay reverse tran scri p ti on PCR ,DDR T 2PCR ) m RNA DDR T 2PCR 的基本原理是合成两对引物,通过随机组合,使每种m RNA 均有扩增机会,并显示出清晰的电泳带。为达到该目的,利用真核细胞m RNA 均含po ly (A )末端以及po ly (A )上游2个碱基只有12种组合(A T 、A C 、A G 、T T 、TC 、T G 、CT 、 CC 、CG 、GT 、GC 、GG ,即42 个组合除去TA 、CA 、 GA 、AA 4个组合)的特点,设计3’ 引物T 11-12M N (M N 2A 、G 、C 、T 中的任一种,M 不能为T ),分别与总m RNA 进行反转录,从而获得12份c DNA ;再以c DNA 为模板,以上述T 11-12M N 为下游引物,以另一随机寡核苷酸引物(如10m er 随机引物)为上游引物进行PCR 扩增。理论上,5’端引物随机结合在c DNA 上,且每一条c DNA 均有扩增机会,获得大 小不同的扩增片段。扩增时掺入同位素标记的单核苷酸,扩增结束后,经序列分析PA GE 后进行放射自显影,如果模板c DNA 存在差异,即可显示差异表达的DNA 片段,最后可通过回收目的DNA ,经 杂交、克隆、测序进行鉴定。工作流程(见图1。 )图1mRNA DDR T 2PCR 工作流程图 F ig 1Schem atic diagram of mRNA DDR T 2PCR 112 c DNA 示差分析(c DNA 2rep resen tati onal difference analysis ,c DNA 2RDA ) c DNA 2RDA 是HU BAN K 和SCHA T Z (1994)在L IS IT SYN 等(1993)建立的基因组DNA 示差分析技术(rep resen tati onal difference analysis ,RDA )基础上改进而来,它是将差减杂交与PCR 相结合的技术。利用双链DNA 为模板时可通过PCR 呈指数扩增,而单链DNA 为模板时则呈线性扩增的原理,首先制备tester c DNA 和driver c DNA ,并用识别序列为4碱基的限制性核酸内切酶(如Dp n 或Sau 3A )消化成平均长度为256bp 的c DNA 片段。随后接上寡核苷酸接头(adap to r R )并按adap to r R 序列设计引物进行PCR 扩增,使两组的c DNA 得到富集,制备扩增子(am p licon s )。由于Dp n 或Sau 3A 的识别序列(GA TC )较常见,这样设计可确保几 乎所有表达基因至少有一次被扩增机会。然后切除接头,只在tester c DNA 片段的5’端接上新接头,接着将其与大大过量的driver DNA 混合进行变性 免疫学杂志第16卷第4期2000年7月 I M M UNOLO G I CAL JOU RNAL V o l 16N o 4Ju ly 2000 S 111