杂多酸催化剂

Heteropoly acids:a green and e?cient heterogeneous Br ?nsted acidic catalyst for the intermolecular hydroamination of ole?ns
Lei Yang a ,Li-Wen Xu a,b,*,Chun-Gu Xia a,*
a
State Key Laboratory for Oxo Synthesis and Selective Oxidation,Lanzhou Institute of Chemical Physics,Chinese Academy of Sciences,
and Graduate School of the Chinese Academy of Sciences,Lanzhou 730000,PR China
b
Department of Chemistry,National University of Singapore,3Science Drive 3,Singapore 117543,Republic of Singapore
Received 8February 2008;revised 5March 2008;accepted 6March 2008
Available online 10March 2008
Abstract
Intermolecular hydroamination of non-activated ole?ns with amides and benzyl carbamate proceeds e?ciently in the presence of environmentally benign silicotungstic acid (HSiW)catalyst under mild conditions in air to a?ord addition products in good to excellent yields.
ó2008Elsevier Ltd.All rights reserved.
Keywords:Heteropoly acids;Intermolecular hydroamination;Amides;Ole?ns
In recent years,Keggin type heteropoly acids (HPAs)catalysts have received much attentions in both academic and industrial applications due to their unique properties,which o?ers several advantages in terms of catalytic perfor-mance,strong acidic,and redox site and selectivity to par-ticular reaction product by selective stabilization of reaction intermediate.1HPAs are non-corrosive,environ-mentally benign,and economically feasible solid acid cata-lysts compared to conventional homogeneous acids,such as H 2SO 4or TfOH.Furthermore,they can be reused and recycled easily in most cases after the reaction and hence they are regarded as green catalysts.As a consequence,a variety of synthetically useful transformations have been developed using HPAs as catalysts,such as oxidation of alcohols,2esteri?cation,3Friedel–Crafts reactions,4Man-nich reactions,5cyanosilylation,6ring-opening of epox-ides,7and dehydration.8
Hydroamination,the simple addition of an N–H bond across C–C unsaturated organic fragment,has attracted much attention in the past decades.Intermolecular hydro-amination of ole?ns is one of the most important and chal-lenging topics in this area.9Despite signi?cant e?orts that have been devoted into the intermolecular hydroamination of ole?ns with alkylamines and arylamines,only a few reports of the intermolecular hydroamination of non-acti-vated alkenes with weakly basic amine nucleophiles such as sulfonamides,carbamates,and carboxamides are known (Scheme 1).
Recently,e?cient platinum(II),10gold(I),11Cu(II),12Fe(III),13and other metal salts 14catalyzed hydroamina-tions of amides and carbamates were reported.Along with the metal catalysts,there also have been examples using metal-free catalysts for the hydroaminations of ole?ns and amides.15Although some notable progress has been made on the hydroamination reactions of alkenes with
0040-4039/$-see front matter ó2008Elsevier Ltd.All rights reserved.doi:10.1016/j.tetlet.2008.03.034
*
Corresponding authors.Tel.:+8609314968056;fax:+8609318277088(L.-W.X.).
E-mail addresses:licpxulw@https://www.360docs.net/doc/8e6156870.html, (L.-W.Xu),cgxia@https://www.360docs.net/doc/8e6156870.html,
(C.-G.
Xia).
Available online at https://www.360docs.net/doc/8e6156870.html,
Tetrahedron Letters 49(2008)2882–2885
amides in the past two years,there are also some draw-backs on the reported methods,such as using expensive and toxic metals,higher reaction temperature,large excess amounts of ole?ns,and tedious reaction procedures.Addi-tionally,most of the reported methods must be carried out under inert atmosphere.Therefore,the development of a novel,green,and simple catalyst for addition reactions of non-activated alkenes with sulfonamides,carboxamides and carbamates remains an intriguing challenge.
Although silver-exchanged tungstophosphoric acid 16or using HPAs as acid additive 17have been examined for intermolecular hydroamination of alkynes with aromatic amines,to the best of our knowledge,there are no studies and reports of HPAs-catalyzed intermolecular hydroamin-ation of ole?ns with amides.In the present letter,we report an e?ciently,easy operational,and reusable heteropoly acids catalyst for the intermolecular hydroamination of various ole?ns with both amides and carbamates.
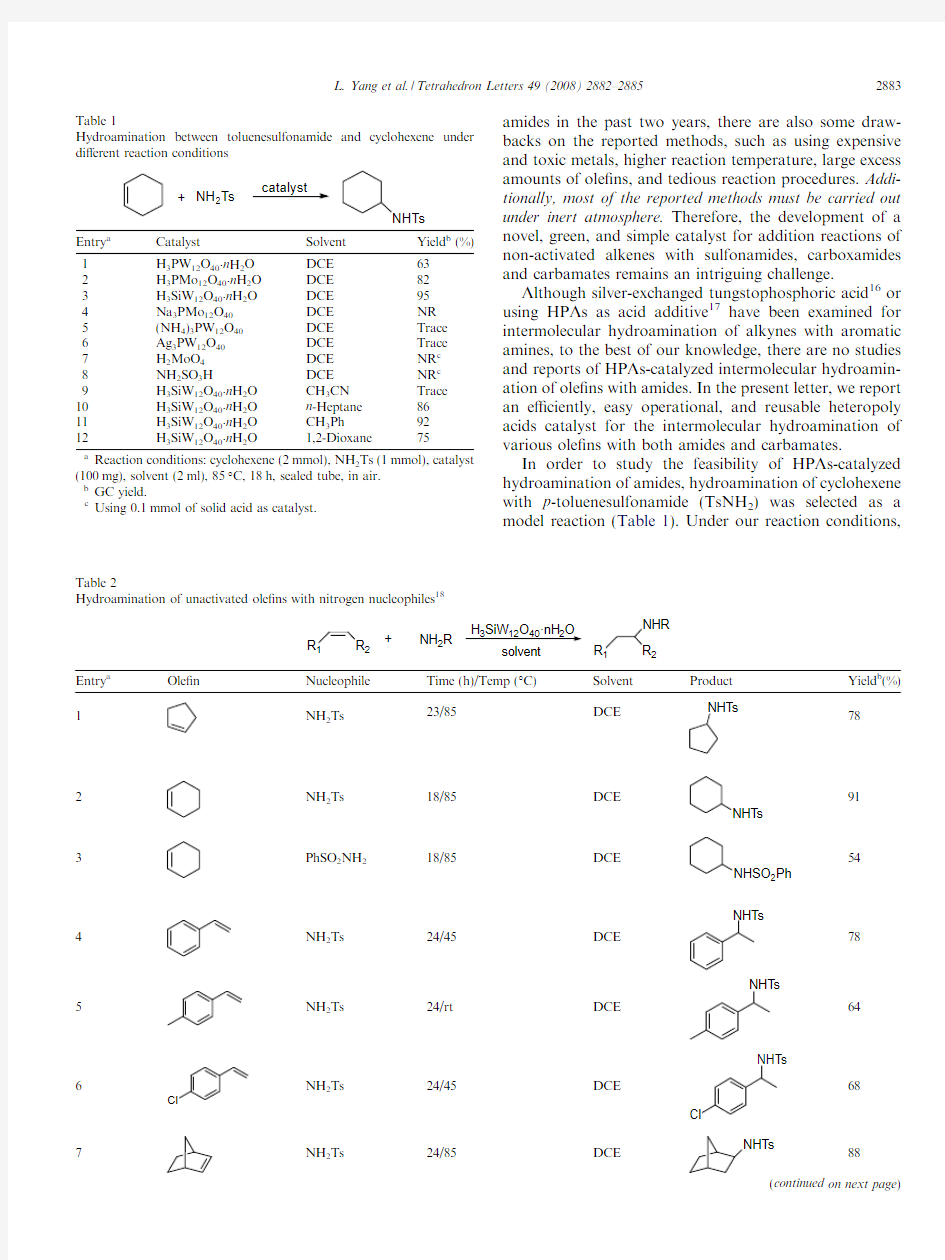
In order to study the feasibility of HPAs-catalyzed hydroamination of amides,hydroamination of cyclohexene with p -toluenesulfonamide (TsNH 2)was selected as a model reaction (Table 1).Under our reaction conditions,
Table 1
Hydroamination between toluenesulfonamide and cyclohexene under di?erent reaction
conditions
+NH 2Ts
NHTs
catalyst
Entry a Catalyst
Solvent Yield b (%)1H 3PW 12O 40án H 2O DCE 632H 3PMo 12O 40án H 2O DCE 823H 3SiW 12O 40án H 2O DCE 954Na 3PMo 12O 40DCE NR 5(NH 4)3PW 12O 40DCE Trace 6Ag 3PW 12O 40DCE Trace 7H 2MoO 4DCE NR c 8NH 2SO 3H
DCE NR c 9H 3SiW 12O 40án H 2O CH 3CN Trace 10H 3SiW 12O 40án H 2O n -Heptane 8611H 3SiW 12O 40án H 2O CH 3Ph
9212
H 3SiW 12O 40án H 2O
1,2-Dioxane
75
a
Reaction conditions:cyclohexene (2mmol),NH 2Ts (1mmol),catalyst (100mg),solvent (2ml),85°C,18h,sealed tube,in air.b
GC yield.c
Using 0.1mmol of solid acid as catalyst.
Table 2
Hydroamination of unactivated ole?ns with nitrogen nucleophiles 18
R 1
2
+
NH 2R
H 3SiW 12O 40·nH 2O
solvent
R 1R 2
NHR Entry a Ole?n
Nucleophile Time (h)/Temp (
°C)Solvent Product
Yield b (%)1
NH 2
Ts
23/85
DCE
NHTs
78
2NH
2Ts 18/85DCE NHTs
91
3
PhSO 2NH 2
18/85
DCE
NHSO 2Ph
54
4
NH 2Ts
24/45
DCE
NHTs
78
5
NH 2Ts
24/rt
DCE
NHTs
64
6
Cl
NH 2Ts 24/45DCE
NHTs
Cl
68
7
NH 2Ts
24/85
DCE
NHTs
88
(continued on next page )
L.Yang et al./Tetrahedron Letters 49(2008)2882–2885
2883
among the HPAs tested,H3SiW12O40án H2O was found to be the most e?cient catalyst compared to H3PW12O40án H2O and H3PMo12O40án H2O.Solid acids such as NH2SO3H and H2MoO4could not furnish the desired products.No or only trace products were found when using the HPAs salts such as Na3PMo12O40, (NH4)3PW12O40,and Ag3PW12O40as https://www.360docs.net/doc/8e6156870.html,ing H3SiW12O40án H2O as catalyst,various solvents were then examined.Among the solvents examined,non-polar DCE was found to give better results in terms of chemical yields. Polar solvent1,4-dioxane tended to shut down the reacting system and a?orded the addition product in lower yield.
To investigate the scope of the H3SiW12O40án H2O cata-lyzed hydroamination reaction,several selected ole?ns and amides were then examined under given reaction condi-tions and the results are summarized in Table2.17Cyclo-pentene and cyclohexene were successfully converted to the corresponding N-cyclopentenyl p-toluenesulfonamide and N-cyclohexyl p-toluenesulfonamide.PhSO2NH2gave lower yield of the addition products.The hydroamination reactions of NH2Ts with various vinyl arenes gave fast and high-yielding reactions under mild reaction conditions. Interestingly,addition of NH2Ts to p-methylphenylene cat-alyzed by H3SiW12O40án H2O occurred even at room tem-perature in good yield.Similarly,the additions of various amides to the more strained norbornene furnished the hydroamination products in good to excellent yields under modi?ed reaction conditions.
To further extend the scope of the substrate,1,3-cyclo-hexadiene was investigated in the reaction of amides and NH2Cbz.All the nitrogen donors underwent hydroamina-tion with good yields at50°C within24–26h.The possibil-ity of the recycling of the catalyst was also examined.For this reaction,the hydroamination of NH2Ts and styrene in DCE at45°C in the presence of catalytic H3SiW12O40án H2O was selected as a model.When the reaction com-pleted,the catalyst could be recycled after simple?ltration and washed with DCE.The obtained powder was dried under vacuum,and then directly reused in subsequently recycled reaction.After two runs,the yield of the corre-sponding addition compound was somewhat decreased (74%,62%).
In conclusion,we have developed a commercially avail-able and reusable HPAs catalyzed intermolecular hydro-amination of unactivated alkenes with weakly basic amine nucleophiles such as sulfonamides,benzyl carba-mate,and benzamide,which can be used successfully and environmental friendly to a?ord good to excellent yields of addition products.Furthermore,this methodology o?ers signi?cant improvements with regard to the scope of this transformation,simplicity in operation,and green aspects by avoiding toxic metals or corrosive catalysts. Acknowledgments
This study was supported by the National Natural Science Founder of China(Project No.20572114)and National Natural Science Funds for Distinguished Young Scholar,PR China(Project No.20625308).
Table2(continued)
Entry a Ole?n Nucleophile Time(h)/Temp(°C)Solvent Product Yield b(%)
8CH3NHTs24/85DCE NMeTs
60
9PhCONH228/85Dioxane NHCOPh
67
10NH2Ns28/85Dioxane NHNs
85
11PhSO2NH228/85DCE NHSO2Ph
86
12NH2Ts24/50DCE NHTs
62
13PhSO2NH224/50DCE NHSO2Ph
48
a Reaction conditions:ole?n(2mmol),NH
2Ts(1mmol),H3SiW12O40án H2O(100mg),solvent(2ml),sealed tube,in air.
b Isolated yield.
2884L.Yang et al./Tetrahedron Letters49(2008)2882–2885
References and notes
1.(a)Mizuno,N.;Misono,M.Chem.Rev.1998,98,199;(b)Timofeeva,
M.N.Appl.Catal.A2003,256,19;(c)Firouzabadi,B.;Iranoor,N.;
Jafari,A.A.Synlett2005,299.
2.Firouzabadi,H.;Iranpoor,N.;Amani,K.Synthesis2003,408.
3.Das,J.;Parida,K.M.J.Mol.Catal.A:Chem.2007,264,248.
4.(a)Kaur,J.;Gri?n,K.;Harrison,B.;Kozhevnikov,I.V.J.Catal.
2002,208,448;(b)Lan,K.;Fen,S.;Shan,Z.-X.Aust.J.Chem.2007, 60,80.
5.(a)Azizi,N.;Torkiyan,L.;Saidi,https://www.360docs.net/doc/8e6156870.html,.Lett.2006,8,2079;(b)
Rasalkar,M.S.;Bhilare,S.V.;Deorukhkar,A.R.;Darvatkar,N.B.;
Salunkhe,M.M.Can.J.Chem.2007,85,77;(c)Wang,E.;Huang,T.-K.;Shi,L.;Li,B.-G.;Lu,X.-X.Synlett2007,2197.
6.Firouzabadi,H.;Iranpoor,N.;Jafari,https://www.360docs.net/doc/8e6156870.html,anomet.Chem.
2005,690,1556.
7.Azizi,N.;Saidi,M.R.Tetrahedron2007,63,888.
8.Dias,A.S.;Lima,S.;Pillinger,M.;Valente,A.A.Carbohydr.Res.
2006,341,2946.
9.(a)Bytschkov,I.;Doye,https://www.360docs.net/doc/8e6156870.html,.Chem.2003,935;(b)Xu,L.
W.;Xia,https://www.360docs.net/doc/8e6156870.html,.Chem.2005,633;(c)Hultzsch,C..Adv.
Synth.Catal.2005,347,367;(d)Matsunaga,https://www.360docs.net/doc/8e6156870.html,.Chem.
Jpn.2006,64,778.
10.(a)Qian,H.;Widenhoefer,R. https://www.360docs.net/doc/8e6156870.html,.Lett.2005,7,2635;(b)
Karshtedt,D.;Bell,A.T.;Tilley,T.D.J.Am.Chem.Soc.2005,127, 12640.
11.(a)Zhang,J.-L.;Yang,C.-G.;He,C.J.Am.Chem.Soc.2006,128,
1798;(b)Liu,X.;Li,C.;Che,https://www.360docs.net/doc/8e6156870.html,.Lett.2006,8,2707;(c)Bender,
C.F.;Widenhoefer,https://www.360docs.net/doc/8e6156870.html,.Lett.2006,8,5303;(d)Brouwer,C.;
He,C.Angew.Chem.,Int.Ed.2006,45,1744.
12.Taylor,J.G.;Whittall,N.;Hii,https://www.360docs.net/doc/8e6156870.html,.Lett.2006,8,3561.
13.Michaux,J.;Terrasson,V.;Marque,S.P.;Wehbe,D.;Campagne,J.
https://www.360docs.net/doc/8e6156870.html,.Chem.2007,2601.
14.(a)Huang,J.;Wong,C.M.;Xu,F.;Loh,T.P.Tetrahedron Lett.
2007,48,3375;(b)Qin,H.;Yamagiwa,N.;Matsunaga,S.;Shibasaki, https://www.360docs.net/doc/8e6156870.html,n J.2007,2,150;(c)Qin,H.;Yamagiwa,N.;
Matsunaga,S.;Shibasaki,M.J.Am.Chem.Soc.2006,128,1611. 15.(a)Talluri,S.K.;Sudalai,https://www.360docs.net/doc/8e6156870.html,.Lett.2005,7,855;(b)Li,Z.;
Zhang,J.;Brouwer,C.;Yang,C.-G.;Reich,N.W.;He,https://www.360docs.net/doc/8e6156870.html,.Lett.
2006,8,4175;(c)Rosenfeld, D. C.;Shekhar,S.;Takemiya, A.;
Utsunomiya,M.;Hartwig,J. https://www.360docs.net/doc/8e6156870.html,.Lett.2006,8,4179;(d) Motokura,K.;Nakagiri,N.;Mori,K.;Mizugaki,T.;Ebitani,K.;
Jitsukawa,K.;Kaneda,https://www.360docs.net/doc/8e6156870.html,.Lett.2006,8,4617.Selected examples of hydroamination with amines catalyzed by Br?nsted acids:(a)
Schlummer,B.;Hartwig,https://www.360docs.net/doc/8e6156870.html,.Lett.2002,4,1471;(b)Anderson, L.L.;Arnold,J.;Bergman,R.G.J.Am.Chem.Soc.2005,127, 14542;(c)Marcsekova′,I.;Doye,S.Synthesis2007,145;(d) Ackermann,L.;Kaspar,L.T.;Althammer,https://www.360docs.net/doc/8e6156870.html,.Biomol.Chem.
2007,5,1975.
16.Lingaiah,N.;Seshu Babu,N.;Mohan Reddy,K.;Sai Prasad,P.S.;
Suryanarayana,https://www.360docs.net/doc/8e6156870.html,mun.2007,278.
17.Mizushima,E.;Hayashi,T.;Tanaka,https://www.360docs.net/doc/8e6156870.html,.Lett.2003,5,3349.
18.A typical procedure for intermolecular addition reactions of amides or
carbamates to ole?ns:Into a test tube were placed H3SiW12O40án H2O (100mg),NH2Ts(1mmol),CH2ClCH2Cl(2ml),and cyclohexene (2mmol).After sealing,the reaction mixture was heated at85°C and stirred vigorously for18h.After the reaction completed,the mixture was concentrated in vacuo to remove the DCE.The product was puri?ed by column chromatography(ethyl acetate/petroleum=1/10–1/5)to gain the analytically pure product(91%isolated yield).All the known compounds were determined by GC–MS or NMR.The spectral data of some representative products are given below.
N-Cyclohexyl p-toluenesulfonamide(Table2,entry2):1H NMR (400MHz)d7.77(d,J=8.0Hz,2H),7.28–7.26(d,J=8.0Hz,2H),
4.55(d,J=6.4Hz,1H),3.10–3.07(m,1H),2.40(s,3H),1.73–1.70
(m,2H),1.62–1.58(m,2H),1.50–1.46(m,1H),1.29–1.06(m,5H).13C NMR(100MHz)d143.09,138.40,129.60,126.90,52.53,33.89,25.10,
24.60,21.51.GC–MS,m/z253.
N-Cyclopentyl p-toluenesulfonamide(Table2,entry1):1H NMR (400MHz)d7.75(d,J=8.0Hz,2H),7.26(d,J=15.6Hz,2H),4.96 (s,1H),3.56–3.51(m,1H),2.44(s,3H),1.73–1.70(m,2H),1.64–1.59 (m,2H),1.52–1.46(m,1H),1.25–1.08(m,5H).13C NMR(100MHz) d143.15,137.77,129.59,127.06,55.06,33.30,23.05,21.47.GC–MS, m/z239.
N-exo-bicyclo[2.2.1]hept-2-yl-p-toluenesulfonamide(Table2,entry
7):1H NMR(400MHz)d7.73(d,J=7.2Hz,2H),7.28(d,
J=7.6Hz,2H),4.61(d,J=6.4Hz,1H),3.10(s,1H),2.41(s,3H),
2.16(s,1H),2.07(s,1H),1.59–1.54(m,1H),1.43–1.38(m,2H),1.31–
1.29(m,2H), 1.15–1.03(m,2H),0.99–0.87(m,2H).13C
NMR(100MHz)d143.19,137.84,129.63,127.06,56.61,42.44,
40.74,35.54,35.14,27.96,26.27,21.51.GC–MS,m/z265.
Cyclohex-2-enyl-p-toluenesulonamide(Table2,entry12):1H NMR (400MHz)d7.75(d,J=7.6Hz,2H),7.27(d,J=8.0Hz,2H),5.71 (d,J=10Hz,1H),5.31(d,J=10.4Hz,1H),4.84(d,J=8.4Hz, 1H),3.765(s,1H),2.39(s,3H),1.94–1.77(m,2H),1.74–1.61(m,1H),
1.57–1.50(m,3H).13C NMR(100MHz)d143.14,138.24,131.39,
129.60,126.95,126.91,48.89,30.12,24.38,21.45,19.22.GC–MS,m/z 251.
L.Yang et al./Tetrahedron Letters49(2008)2882–28852885
负载型杂多酸钒催化剂浅析
负载型杂多酸钒催化剂浅析 开封市三丰催化剂有限责任公司 耿雨 张智勇 杜保强 [摘 要] 本研究主要对用于硫酸生产的固体负载杂多酸钒催化剂主要成份、杂多酸与载体的相互作用等问题进行初步探索,针对现有硫酸催化剂的生产工艺作出改进,提高催化剂的活性以及选择更适宜的物化性质(孔容积、孔径分布、比表面、强度、颗粒度和形状等)。 [关键词] 硫酸催化剂 杂多酸 催化剂选择活性 我国钒催化剂品种有中温型、低温型、宽温区型,也有特种耐砷型,外形有条形、环形和异形等。生产工艺有混碾工艺和后期浸渍处理,尽管很多厂家进行了许多改良,但品种基本在这些范畴内,并没有实质性的突破。未从本质上解决催化剂存在的问题,本研究是从催化剂生产原料硅藻土的研究入手,探索杂多酸催化成份在多孔二氧化硅上的分布状态,从而解决硫酸催化剂具体微观组分热稳定性,改善其相应物化性能。 1.二氧化硅载体 目前世界上所有的硫酸生产用钒催化剂载体的有效成份均为硅藻土所含的无定型SiO2,它决定了催化剂的最初强度,理论上认为无定型SiO2不参与反应,但从实际应用情况判断无定型SiO2直接影响有效催化剂成份、SO2与O2的传递(微观反应的扩散过程)。通常在320-610℃下,催化剂的活性组分在二氧化硅载体表面形成了很薄的液膜(100-1000 ?),SO2穿过液膜时与一个独立的钒位表面氧化成SO3时造成了V-O-M键的变化(M为K或P或Cs或Mn或Li或Ti 等),由于M氧化物具有多变性,SO2在多变的负载钒催化剂上的催化活性也呈现多变性,这就需要稳定的SiO2载体来提高硫酸催化剂的稳定性。 1.1载体塑性 硅藻土孔隙中所含的是由吸附水和结合水组成,其中结合水是在双电层范围内的被土颗粒吸引在其周围的水,它分为强结合水和弱结合水;吸附水是处于土颗粒引力范围之外的水,它分为重力水和毛细水,自由水在硫酸催化剂中需110℃干燥1.5h才能逸出,而结合水需在450℃下连续焙烧1.5小时。 I p来表示。 固态 半固态 可塑态 液态 图1-1 硅藻土的界限含水量 137
杂多酸
液体催化剂制备技术及应用 赵毓璋
1. 研发杂多酸催化剂的意义 催化剂的应用历史很长,特别在石油化工、精细化工、有机化工和生物化工中,可以说催化技术已成为化学工业最关键的核心技术之一。据统计,到目前为止,人类所掌握的化学反应80%以上必须在催化剂存在下才能实现。在化学工业生产中,最常用的催化剂是无机酸和无机碱,一般是液体溶液,用于均相或非均相反应。酸碱催化剂适用于水合反应、分解反应、酯化反应、芳烃烷基化反应、脱水反应、胺化反应、加氢反应、不饱和化合物的双键转移反应、氧化还原反应等。但是由于传统的酸碱催化剂过于注重生产的实效性和经济性,而忽略环境效应和生态效应,以至于目前所使用的催化剂绝大多数都对环境造成或多或少的污染。如今有害化学物质的处理和环境保护受到特别关注,世界各国都在积极进行绿色化学研究与开发,提倡清洁生产,特别是化学化工中的清洁生产更为世人瞩目,它已成为主要的研究方向。绿色化学是更高层次的化学,它的主要特点是原子经济,即在获取新物质的转化过程中充分利用每个原料原子,实现“零排放”,既可充分利用资源,又不产生污染,实现清洁生产。而催化技术是清洁生产的重要技术,因此,研究和开发新的环境友好型催化剂是摆在科学工作者面前的一个比较迫切的课题。目前,这方面的研究有固体超强碱催化剂、杂多酸催化剂、夹层式催化剂等的开发。 杂多酸(Heteropoly Acid,简称HPA )是由杂原子(如P、Si、Fe、Co等)和多原子(如Mo、W、V、Nb、Ta等)按一定的结构
通过氧原子配位桥联组成的一类含氧多酸,具有很高的催化活性,它不但具有强酸性,而且具有氧化还原性,是一种多功能的新型催化剂。杂多酸稳定性好,可作均相及非均相反应,甚至可作相转移催化剂,对环境无污染,是一类大有前途的绿色催化剂,它可用作以芳烃烷基化和脱烷基反应、酯化反应、脱水/化合反应、氧化还原反应以及开环、缩合、加成和醚化反应等。因杂多酸独特的酸性、“准液相”行为、多功能等优点在催化反应领域已有许多出色的应用实例。如丙烯液相水合制异丙醇、甲基丙烯醛氧化制甲基丙烯酸、四氢呋哺(THF)开环聚合加水合制聚氧四甲撑二醇(PTMG)它是合成聚氨酯的主要原料。杂多酸在均相、多相酸催化反应、氧化还原反应中都有许多别于其它催化材料的特性,概括有如下几点: (1)杂多酸结构组成简单、性能稳定,其催化性能容易用杂多酸阴离子的分子水平表征。 (2)杂多酸的表面结构和体相结构差别很小,具有所谓“准液相”的特征。催化反应不仅在表面上进行,同时在体相内进行。 (3)杂多酸不仅同时具有多元酸如多电子还原能力,而且它的酸性和氧化还原性还可以在较大的范围内调变。因此,杂多酸即可作为酸性催化剂又可作为氧化还原催化剂,是一种双功能催化剂。 (4)杂多酸具有较好的热稳定性和可溶性,因此它既可作为多相催化剂,也可作为均相催化剂。 (5)杂多酸的酸强度远远高于通常的无机酸,但是由于质子不游离出来,腐蚀性很小。
杂多酸催化剂
题目杂多酸催化剂 姓名与学号张凌烽 1108010236 指导教师孟锦宏 年级与专业2011级化学工程与工艺 所在学院环境与化学工程学院
摘要 杂多酸是固体酸的一种,具有着独特的氧化还原性,酸性以及双功能性,在许多化学反应中能够表现出很强的催化活性。杂多酸这种绿色、无毒、无腐蚀性的环保型催化剂己在多种有机反应中实现了成功应用,如:酯化醚化、缩合反应、酰基化、烷基化、水合脱水和聚合反应等,反应中呈现出反应活性高、腐蚀性小、污染率低等诸多优点,但由于杂多酸比表面积小、热稳定性低、回收困难等问题使得杂多酸在催化领域的应用受到了一定的限制。 关键词:
1 绪论 1.1前言 绿色化学是近十年来在化学领域内提出的新名词,绿色化学又被称为“环境友好化学“、“清洁化学”、“环境无害化学”。这种发展趋势已涉及到分子合成、生物技术、化学分析等许多领域,内容丰富,应用广泛。绿色化学的最大优势在于通过科学的手段在化学反应的起始与末端进行有效的防控干预,使反应中无副产物,真正实现零排放,彻底无污染,化学绿色化是新时代里化学发展的主要研究方向。 无机酸是许多化工产品生产中必不可少的、非常重要的常规催化剂,传统无机酸类催化剂主要有浓硫酸、三氯化招、浓憐酸等。这类酸催化剂在反应中有许多优势,如工艺成熟、催化效率高、价格低廉,但此类催化剂最大的缺陷在于:副反应多、腐烛性强、设备要求高、后处理繁杂,无法满足环保技术的要求,为了克服诸多缺点,人们开发、研制了许多新型催化剂,如固体酸、杂多酸和离子交换树脂等。 杂多酸是一类含有氧桥的多核高分子化合物,无论是均相反应体系,还是非匀相反应体系,杂多酸均可作为酸催化剂,氧化还原型催化剂及双功能型催化剂,广泛应用于各类有机反应催化当中,如:酯化酸化、缩合反应、酰基化、烧基化、水合脱水、聚合反应等,反应中呈现出腐烛性小,活性高,污染率低等诸多优点。二十世纪七十年代,憐鹤酸催化丙稀水合制备异丙醇在R本成功投入工业生产。目前,以杂多酸为催化媒介并实现工业化生产的的重要有机合成反应已达十几种随着科学研究的不断开拓深入,杂多酸类化合物在工业催化领域的开发将越来越深入。 1.2多酸化学简介 时至今日,多酸化学的发展己有200年的历史,进入新世纪后,多酸化学走进了一个薪新的发展时代“?”。多酸分子中的金属离子通常具有d"电子构型,最具代表性的是鹤原子和销原子,也是构成多酸的主要元素。多酸的立体结构中,八面体
环己酮催化缩合可能用到的催化剂总结
合成可能用催化剂总结: 1、硫酸氢钾,熔点197℃,暂时符合反应温度,弱酸性。还可考虑与三 氧化二铝同时应用,催化效果更加明显。 2、碳酸钠,400℃分解,弱碱性,符合温度及反应条件,且不腐蚀设备, 无污染。 3、固体超强碱,此为研究热点,有多种,如下: (1)Na|NaOHγ-AL2O3,反应温度190℃,符合气相催化温度要求,且活性高,重点考虑。 (2)Na-Na2CO3γ| AL2O3,查阅文献得到的超强碱,无法确定能否使用。 (3)CaO|ZrO2-La2O3固体碱,此碱可合成酯,考虑到反应与酮缩合有差异,待定。 (4)几种三氧化二铝固体超强碱,这些催化剂不是专门合成2-(1-环己烯基)环己酮的,但可以考虑,KF\γ-AL2O3,KNO3\ AL2O3, K2CO3\ AL2O3,Na-KOH\γ-AL2O3。 (5)另外所查几种固体碱催化剂,KOH/La203-Mg0,La203-ZrO2,Ca0/Zr02-La203,Na2Sn03,MgO-Sn02,Na-KOH-Mg0。 (6)还有分子筛型固体碱催化剂,2Na+02-/Al-MCM-41固体超强碱等。 4、固体超强酸,多种,如下: (1)SO42-\M X O Y,此催化剂催化缩酮反应反应温度在160℃,较符合。 (2)S042-/Ti02-Al-MCM-41型分子筛固体超强酸催化剂,5042"/Ti02-Sn02-Al-MCM-41分子筛型固体超强酸催化剂。 (3)纳米SO42-\SnO2固体超强酸,已有对缩酮反应的研究,温度较符合。 (4)铁系新型固体超强酸Fe203/S2082-/La 3+,目前已有其对环己酮缩乙二醇的合成研究,温度对本反应不太符合。 (5)几项专利,其一,SO42-/M x O y型固体超强酸具有无卤素离子,无污染无腐蚀,易与反应物分离,以及能在高温仍然保持活性 和稳定性等优点;其二,固体超强酸催化剂SO2 -4/TiO2 WO3 , 并以丁酸丁酯的合成作为探针反应,系统考察了WO3 的含 量、硫酸浸渍浓度、焙烧温度等制备条件对SO2 -4/TiO2 WO3 催化活性的影响;其三,用sol-gel法合成了纳米KF/Al2O3 超强碱催化剂,用均匀设计软件研究了其在Knoevenagel缩合 和Michael加成反应中的应用。
纳米催化剂简介
纳米催化剂简介 摘要 催化剂的作用主要可归结为三个方面:一是提高反应速度,增加反应效率;二是决定反应路径,有优良的选择性,例如只进行氢化、脱氢反应,不发生氢化分解和脱水反应;三是降低反应温度。纳米粒子作为催化剂必须满足上述的条件。近年来科学工作者在纳米微粒催化剂的研究方面已取得一些结果,显示了纳米粒子催化剂的优越性。 纳米微粒由于尺寸小,表面所占的体积百分数大,表面的键态和电子态与颗粒内部不同,表面原子配位不全等导致表面的活性位置增加,这就使它具备了作为催化剂的基本条件。最近,关于纳米微粒表面形态的研究指出,随着粒径的减小,表面光滑程度变差,形成了凸凹不平的原子台阶,这就增加了化学反应的接触面。有人预计超微粒子催化剂在下一世纪很可能成为催化反应的主要角色。尽管纳米级的催化剂还主要处于实验室阶段,尚未在工业上得到广泛的应用,但是它的应用前途方兴未艾。 关键词:性质,制备,典型催化剂,表征技术,应用,
目录 绪论-----------------------------------------------------------1 1. 纳米催化剂性质----------------------------------------------1 1.1 纳米催化剂的表面效应-------------------------------------1 1.2 体积效应-------------------------------------------------1 1.3 量子尺寸效应---------------------------------------------1 2. 纳米催化剂的制备--------------------------------------------2 2.1 溶胶凝胶法-----------------------------------------------2 2.2 浸渍法---------------------------------------------------2 2.3 沉淀法---------------------------------------------------3 2.4 微乳液法-------------------------------------------------3 2.5 离子交换法-----------------------------------------------3 2.6 水解法---------------------------------------------------3 2.7 等离子体法----------------------------------------------3 2.8 微波合成法-----------------------------------------------4 2.9 纳米材料制备耦合技术-------------------------------------4 3. 几种典型催化剂----------------------------------------------4 3.1 纳米金属粒子催化剂---------------------------------------4 3.2 纳米金属氧化物催化剂-------------------------------------5 3.3 纳米半导体粒子的光催化-----------------------------------5 3.4 纳米固载杂多酸盐催化剂-----------------------------------5 3.5 纳米固体超强酸催化剂-------------------------------------6 3.6 纳米复合固体超强酸催化剂---------------------------------6 3.7 磁性纳米固体酸催化剂-------------------------------------6 3.8 碳纳米管催化剂-------------------------------------------7 3.9 其它纳米催化剂-------------------------------------------7 4. 纳米催化剂表征技术------------------------------------------7
杂多酸的研究进展1108010224李轶凡
摘要 杂多酸(Heteropoly Acid,简写为HPA )是由杂原子(如P、Si、Fe、Co等)和多原子(如Mo、W、V、Nb、Ta等)按一定的结构通过氧原子配位桥联组成的一类含氧多酸,具有很高的催化活性,它不但具有酸性,而且具有氧化还原性,是一种多功能的新型催化剂,杂多酸稳定性好,可作均相及非均相反应,甚至可作相转移催化剂,对环境无污染,是一类大有前途的绿色催化剂,它可用作以芳烃烷基化和脱烷基反应、酯化反应、脱水/化合反应、氧化还原反应以及开环、缩合、加成和醚化反应等。因杂多酸独特的酸性、“准夜相”行为、多功能(酸、氧化、光电催化)等优点在催化研究领域中受到研究者们的广泛重视。 关键词:杂多酸催化多功能
目录 杂多酸催化剂 (3) 一、定义 (3) 二、制备 (4) 2.1Dawson杂多酸制备 (4) 2.1.1 Dawson型磷钼钒杂多酸的合成 (4) 2 .2 Keggin型杂多酸的合成 (4) 2.2.1 Keggin型Ni—Mo—Zr杂多酸盐的合成 (4) 2.3 负载型 P—no—W 杂多酸催化剂的制备 (5) 2.3.1直接负载法 (5) 2.3.2接枝法 (5) 2.3.3密封法 (5) 三.应用 (6) 3.1铈钼锆杂多酸盐的制备及超声降解性能 (6) 3.2二氧化硅负载杂多酸铵催化苯液相硝化反应的研究 (6) 四.负载型杂多酸催化剂的研究进展 (7) 4.1活性炭负载杂多酸催化合成没食子酸甲酯的研究 (7) 4.2介孔材料负载杂多酸催化剂催化乙醇脱水制乙烯 (8) 4.3磷钨杂多酸季铵盐催化脂肪酸甲酯环氧化 (8) 4.4纳米复合杂多酸催化合成草莓酯 (9) 4.5杂多酸(盐) 掺杂TiO2 制备新型复合光催化剂的研究进展 (9) 4.6杂多酸催化合成磷酸单双辛酯的研究 (10) 参考文献 (11)
催化原理总结
第一章 1.催化反应的三个特征 2.催化反应三种分类 催化剂两种分类 3.固体催化剂四种组成 助催化剂四种结构和载体的五种作用 催化剂的微观结构 4.催化反应性能的三个标准 5.多相催化反应的七个步骤 第二章 物理吸附和化学吸附的区别 物理吸附和化学吸附位能曲线 解离吸附和谛合吸附的概念 化学吸附态(H2,O2,CO,烯烃,炔烃) 吸附等温线(五种) Langmuir吸附等温方程推导(单分子,多分子)BET方程推导催化剂比表面 测定比表面的方法(N2吸附) 催化剂密度(真密度,假密度,比孔容,孔隙率,平均孔半径,孔径分布) 扩散(Kundsen扩散) 第三章 3.1酸碱催化剂的应用和分类(了解) 3.2酸碱定义及酸碱中心的形成 L酸和B酸定义; 单元氧化物L酸吸水转化为B酸 Tababe二元氧化物酸中心形成机理(Ti6Si4) 3.3固体酸性质及其测定 H0的定义(H0=pKa+log(CB/CBH+) TPD法和IR法测定酸中心 固体超强酸(H0<-10.6) 3.4 酸碱催化作用及其催化机理 特殊酸碱催化(反应的速控步) Bronsted规则ka=GaKaα 碳正离子的形成和反应规律 酸中心类型与活性选择性的关系 3.5 沸石分子筛 四个结构;四个类型分子筛 沸石分子筛酸中心形成的四种机理 沸石分子筛的择型催化作用及影响因素 催化裂化与热裂化的区别 第四章
4.1 金属催化剂的应用及特性(具有d带电子的元素) 4.2 金属催化剂的化学吸附 以d电子解释金属催化剂的吸附热 (未结合d电子数越大,吸附热大,强吸附Fe; 未结合d电子数少,吸附热小,弱吸附Pt) 2.电子逸出功,电离势概念;两者与化学吸附的关系 3.化学吸附和催化活性的关系 (中等强度的吸附)88页图4-2 4.3金属催化剂电子因素与催化作用的关系 能带理论(金属原子紧密堆积,原子轨道重叠, 分立的电子能级扩展为能带) d带空穴(可以由磁距测得) d带空穴需要适应反应电子数目的转移 价键理论;金属原子的共价键由杂化轨道构成 d%:d轨道占有杂化轨道的比例( Ni,30%d2SP3;70%,d3SP2) d%越大,d带空穴越少; d%增大,吸附热变小,吸附强度变低。 4.4金属催化剂晶体结构与催化作用的关系 晶格,晶格参数和晶面的概念 晶体对催化作用的影响 几何角度:吸附位,键长和键角 能量角度:q=s/2 晶格缺陷引起催化活性变化(了解) 4.5负载型催化剂 分散度的概念;分散度与晶粒大小的关系 结构敏感反应(C-C,C-O)结构非敏感反应(C-H) 金属与载体的相互作用(实验证明的思路) 溢流现象及其应用 4.6合金催化剂 几何效应和电子效应对催化反应活性的影响。 (Ni-Cu和Pd-Au) 4.7金属催化剂催化作用的典型剖析 合成氨催化剂的助剂(Al2O3,K2O,CaO等) Ag为乙烯环氧化反应催化剂的机理 催化重整反应对双功能催化剂的要求 第五章 1.过渡金属氧化物催化剂的结构类型,应用及其特点 2.导体、半导体、绝缘体的能带结构和类型 3.半导体、n型半导体、P型半导体形成 4.杂质对半导体催化剂费米能级Ef、逸出功、和电导率的影响 5.杂质对半导体催化剂的影响 6.半导体催化剂化学吸附与催化作用 A)受电子气体吸附 n型半导体上吸附
固体超强酸制备
探究思路:两个要求:“保证活性高作为前提,以使用次数作为重要比较指标” 其实,一个固定酯化反应采用不同的固体超强酸(均以该酯化反应作为探究优化制备条件)作为催化剂,所得到的酯化效率差别不会大,只要肯花功夫、时间探究便可达到,所以探究重点摆在对比固体超强酸的稳定性上即提高其使用寿命,而使用寿命以催化活性高作为前提(不同催化剂间催化效用相差不大下,尽管催化效率较差点,但使用次数好,这也算是好催化剂),但在催化效用有一定情况下,探究使用寿命才有意义,随意首先需要探究出优化的固体超强酸的制备条件和酯化条件。 借助微波酯化反应探究最佳活性的催化剂制备条件,然后以活性最佳的催化剂探究微波酯化反应条件。 微波辐射酯化反应——“微波辐射催化合成乙酸正丁酯”: 用微波辐射技术以乙酸和正丁醇为原料,S2O2-8/M X O Y型固体超强酸为催化剂的酯化反应,最佳的微波合成条件为:催化剂用量2。0 g,酸醇物质的量的比为1。0∶2。0,微波功率为595 W,微波辐射时间为30 min,产率84。1%。 主要试剂和仪器:冰醋酸(CP),正丁醇(AR),微波炉,阿贝折光仪(或红外光谱波峰测试)实验过程: 在100 mL圆底烧瓶中加入5。7 mL(0。1 mol·L-1)的冰醋酸和9。1 mL(0。1 mol·L-1)的正丁醇(最适宜的酸醇比为1。0∶2。0),加入2。0 g催化剂,然后将圆底烧瓶装好回流冷凝管和搅拌装置,置于微波炉内。在搅拌下先以65 W的功率加热1 min,再以最适宜的微波功率是595 W,一定反应时间加热回流时间30 min。反应完毕取出圆底烧瓶,待反应物稍冷,过滤出催化剂,粗产品经提纯、干燥、蒸馏,收集124~126℃的馏分。称重,计算产率。 在合成反应中,有些反应是可逆反应生成水,为了提高转化率,常用带水剂把水从反应体系中分离出来。可作带水剂的物质必须要与水水作用产生共沸物使得水更易被蒸出,且在水中的溶解度很小.它可以是反应物或者产物,例如如:环已烯合成是利用产物与水形成共沸物;乙酸异戊酯合成中,反应初期利用原料异戊醇与水形成二元共沸物或原料,产物和水形成三元共沸物,并用分水器分水,同时将原料送回反应体系,随着反应的进行,原料减少,则利用产物乙酸异戊酯与水形成 二元共沸物. 带水剂也可以是外加的。反应物及产物沸点比水高但反应又产生水的,外加第三组分,但第三组分必需是对反应物和产物不起反应的物质,通常加入的第三组分有石油醚,苯甲苯,环已烷,氯仿,四氯化碳等。 在250mL单口平底烧瓶中加入10mL正丁醇、6mL乙酸,再加入适量的三氯化铁作催化剂,放入微波炉内,装上回流冷凝管及分水器,在一定功率微波连续辐射后停止反应。冷却至室温,用饱和食盐水洗涤,分出有机层,水洗至中性,用无水硫酸镁干燥,蒸馏,收集124℃~126℃的馏分,
固体超强酸系列催化剂制备
1. 稀土固体超强酸S2O82- / Sb2O3 / La3+催化剂制备: 将8g SbC13溶于40mL乙醇和20mL苯的混合液中,搅拌充分溶解后得透明锑醇液,再向溶液中加入10mL异丙醇,使醇化反应进行得更彻底,然后加入少量阴离子表面活性剂,并滴加氨水,使之发生水解反应,得到胶状沉淀,低温化12h左右,多次洗涤至无Cl-检出。滤饼于110℃烘干后,研磨过100目筛。搅拌下将Sb2O3浸渍在一定浓度的(NH4)2S2O8溶液中lh,用量为每克Sb2O3用15mL(NH4)2S2O8溶液,抽滤,烘干,置于马弗炉中焙烧,得S2O82-/ Sb203催化剂。将Sb2O3浸渍在一定浓度的(NH4)2S2O8和一定浓度的La(NO3)3的混合液1h,抽滤、烘干置于马弗炉在不同的温度和时间下焙烧,得一系列S2O82-/ Sb2O3 / La3+固体超强酸催化剂,置于干燥器中备用。以代号表示不同制备条件下所得催化剂。 参考文献:稀土固体超强酸S2O82- / Sb2O3 / La3+的制备及催化性能研究 舒华1,连亨池2,闫鹏2,文胜2,郭海福2 (1.学院生化系,554300;2.学院化学化工学院,526061) 稀土,2008.12(29卷第6期) 2. 稀土固体超强酸SO42-/TiO2-La2O3制备: 将一定量La203溶于浓度为3.0 mol·L-1的稀盐酸中,配成La3+溶液,再按一定量比量取TiC14与La3+溶液混合,用NH4·H 0[ w(NH3)=12%]水解至溶液呈碱性,控制pH值在8~9,沉淀完全,静置24 h后进行抽滤,并用蒸馏水不断洗涤至沉淀无Cl-存在(用0.1 mol·L-1的AgNO3检验),于105℃烘干后研细.再将该粉末浸泡于浓度为0.8 mol·L-1的稀H2SO4中24 h,然后抽滤,放入干燥箱中在110℃烘干,于一定的温度下焙烧活化3 h,冷却后置于干燥器中备用。 参考文献:稀土改性固体超强酸催化剂SO42-/TiO2-La2 O3的制备及其催化性能 水金,黄永葵,白爱民,赘,聚堂
固体酸催化剂研究近况综述
试卷( A 卷) 专业: 课程代码: 19060071 学号: 姓名: 作文题(任选一题,写一篇综述论文,每题 100 分) 自拟题目,写一篇关于工业上绿色环保催化剂进展的综述论文 [能力层次: 综合运用和创见 ];[难易度: 较难 ] 要求: 1、查阅文献至少在20篇以上,并且外文文献引用2篇以上; 2、论文字数3000字以上; 3、论文格式严格按照综述论文要求书写; 绿色固体酸催化剂研究近况综述 摘 要:催化剂的研究和发展是现代化学工业的核心问题之一,现代化学工业的巨大成就是同使用催化剂联系在一起的。目前90%以上的化工产品,是借助催化剂生产出来的。工业催化的发展是紧随化学工业的演变而发展的。 催化剂和催化技术的研究与应用,对国名经济的许多重要部门是至关重要的。但就化工工艺过程来说,催化剂的应用可以具体概括为以下几个方面:更新原料路线,采用更廉价的原料;革新工艺流程,促进工艺过程的开发;缓和工艺操作条件,达到节能降耗的目的;开发新产品,提高产品收率,改善产品的质量;消除环境污染或开发从原来到产品的整个化工品过程,对资源的有效利用以及污染控制的环境友好的“绿色催化工艺”等。 引言:固体酸催化剂因其具有对多种化学反应有较高活性与选择性、回收重复利用效率较高等优点,已作为绿色环境友好型催化材料备受人们关注。本文主要综述了近年来国内外对各类型固体超强酸、杂多酸固体酸、离子交换树脂的研究近况,并提出了对今后固体酸催化剂发展的展望。 关键词:固体酸;催化剂 【正文】以往单纯追求眼前效益、罔顾环境所造成的危害近年来逐渐得到人们的重视。随着环保意识的增强,以及绿色化学的提出,越来越多的学者致力于开发效益兼顾环境、使化学工业促可持续发展的新型催化剂。催化剂在工业化生产上起着加速反应进行和提高产率的重要作用,其中酸催化剂在催化领域中得到了广
固体超酸及其应用研究进展
固体超酸及其应用研究进展 摘要:目前已制备的超酸种类繁多, 它具有极强的酸性和高介电常数, 在化学合成工业中是一种良好的催化剂。本文对超强酸的定义、酸度的测定进行了简单介绍。固体超强酸是近年来发展的一种新型催化材料,进一步综述了固体超强酸的分类、制备方法,例举了一些学者制备的新的固体超强酸催化剂。重点是介绍固体超强酸催化剂在有机化学反应中的应用。指出了固体超强酸的优点和一些不足。最后指出了今后固体超强酸催化剂的发展方向。 关键词:超酸;固体超酸;催化剂;应用;发展 Abstract: The acid has been prepared over a wide range, it has a very strong acid and high dielectric constant, it is a good catalyst in the chemical synthesis industry.In this paper, the definition of super acid, acidity determination for a brief introduction. Solid superacid is a new type of catalytic material in recent years.the classification of solid superacids and preparation methods are described.New solid superacid catalysts are introduced. solid superacid catalysts are applied in organic reactions which is the key in the article.Pointing out the advantages of solid superacids and some shortcomings. Finally,development trends of solid superacid catalysts are put forward. Key words:Superacid; solid superacid; catalyst; application; development
杂多酸催化剂
Heteropoly acids:a green and e?cient heterogeneous Br ?nsted acidic catalyst for the intermolecular hydroamination of ole?ns Lei Yang a ,Li-Wen Xu a,b,*,Chun-Gu Xia a,* a State Key Laboratory for Oxo Synthesis and Selective Oxidation,Lanzhou Institute of Chemical Physics,Chinese Academy of Sciences, and Graduate School of the Chinese Academy of Sciences,Lanzhou 730000,PR China b Department of Chemistry,National University of Singapore,3Science Drive 3,Singapore 117543,Republic of Singapore Received 8February 2008;revised 5March 2008;accepted 6March 2008 Available online 10March 2008 Abstract Intermolecular hydroamination of non-activated ole?ns with amides and benzyl carbamate proceeds e?ciently in the presence of environmentally benign silicotungstic acid (HSiW)catalyst under mild conditions in air to a?ord addition products in good to excellent yields. ó2008Elsevier Ltd.All rights reserved. Keywords:Heteropoly acids;Intermolecular hydroamination;Amides;Ole?ns In recent years,Keggin type heteropoly acids (HPAs)catalysts have received much attentions in both academic and industrial applications due to their unique properties,which o?ers several advantages in terms of catalytic perfor-mance,strong acidic,and redox site and selectivity to par-ticular reaction product by selective stabilization of reaction intermediate.1HPAs are non-corrosive,environ-mentally benign,and economically feasible solid acid cata-lysts compared to conventional homogeneous acids,such as H 2SO 4or TfOH.Furthermore,they can be reused and recycled easily in most cases after the reaction and hence they are regarded as green catalysts.As a consequence,a variety of synthetically useful transformations have been developed using HPAs as catalysts,such as oxidation of alcohols,2esteri?cation,3Friedel–Crafts reactions,4Man-nich reactions,5cyanosilylation,6ring-opening of epox-ides,7and dehydration.8 Hydroamination,the simple addition of an N–H bond across C–C unsaturated organic fragment,has attracted much attention in the past decades.Intermolecular hydro-amination of ole?ns is one of the most important and chal-lenging topics in this area.9Despite signi?cant e?orts that have been devoted into the intermolecular hydroamination of ole?ns with alkylamines and arylamines,only a few reports of the intermolecular hydroamination of non-acti-vated alkenes with weakly basic amine nucleophiles such as sulfonamides,carbamates,and carboxamides are known (Scheme 1). Recently,e?cient platinum(II),10gold(I),11Cu(II),12Fe(III),13and other metal salts 14catalyzed hydroamina-tions of amides and carbamates were reported.Along with the metal catalysts,there also have been examples using metal-free catalysts for the hydroaminations of ole?ns and amides.15Although some notable progress has been made on the hydroamination reactions of alkenes with 0040-4039/$-see front matter ó2008Elsevier Ltd.All rights reserved.doi:10.1016/j.tetlet.2008.03.034 * Corresponding authors.Tel.:+8609314968056;fax:+8609318277088(L.-W.X.). E-mail addresses:licpxulw@https://www.360docs.net/doc/8e6156870.html, (L.-W.Xu),cgxia@https://www.360docs.net/doc/8e6156870.html, (C.-G. Xia). Available online at https://www.360docs.net/doc/8e6156870.html, Tetrahedron Letters 49(2008)2882–2885
固体超强酸催化剂
论文题目摘要 本文简单介绍了固体超强酸的发展、研究方向、及存在的问题。并采用沉淀-浸渍法制备SO 4/ZrO 2固体超强酸,分别以氧氯化锆和氨水为锆源和沉淀剂,110℃干燥后经硫酸浸渍、干燥和焙烧制得催化剂。同时考察不同焙烧温度对催化剂性能的影响。在最优的条件下用正交设计法考察锆源(Zr(NO 3)4·5H 2O,ZrOCl 2·8H 2O )、沉淀剂(氨水、 乙胺)和硫酸化试剂(硫酸、硫酸铵)对催化剂性能的影响,并通过乙酸和正丁醇的酯化反应的酯化率检测所制备的SO 4/ZrO 2催化剂的催化效果。结果表明采用硫酸浸渍液浓度为0.5mol/L ,焙烧温度为550℃时所制备的催化剂在正丁醇和乙酸的物质的量比为1.3及不添加任何带水剂的密闭条件下酯化效果最好,酯化率为52.5%。正交设计实验中锆源、沉淀剂、硫酸化试剂的极差分析结果分别为1.54%,2.27%,5.11%,此结果说明硫酸化试剂对催化剂的影响最大,其次是沉淀剂,锆源的影响最小,同时表明以Zr(NO 3)4·5H 2O 为锆源、乙胺为沉淀剂、硫酸为硫酸化试剂是催化剂的最佳制备方案。 关键词:SO 4/ZrO 2 ;固体超强酸;正交实验设计;乙酸丁酯
目录 第1章. 前言 (4) 第1.1节固体超强酸催化剂的发展前景 (4) 第1.2节固体超强酸目前存在的问题 (4) 第1.3节固体超强酸的研究方向 (5) 第1.4节本课题的研究意义及方案 (5) 第2章. 实验理论 (6) 第2.1节固体超强酸的酸度定义 (6) 第2.2节固体超强酸的合成方法 (6) 第2.3节催化剂合成原理 (7) 第2.4节催化剂酸性中心结构 (7) 第2.5节酸性中心形成机理 (8) 第2.6节催化剂失活机理 (9) 第2.7节催化剂酯化反应机理 (9) 第2.8节酯化反应原理 (10) 第3章.实验部分 (11) 第3.1节主要仪器与设备 (11) 第3.1.1节主要仪器 (11) 第3.1.2节主要设备 (11) 第3.2节主要试剂与配制 (11) 第3.2.1节主要试剂 (11) 第3.2.2节主要试剂配制 (12) 第3.3节实验内容 (12) 第3.3.1节催化剂的制备 (12) 第3.3.2节酯化反应 (13) 第3.3.3节酯化率的测定 (13) 第3.4节正交实验设计 (14) 第3.4.1节正交实验设计概念 (14) 第3.4.2节本实验正交设计方案 (14) 第3.4.3节实验步骤 (16)
固体超强酸
固体超强酸 百科名片 固体酸克服了液体酸的缺点,具有容易与液相反应体系分离、不腐蚀设备、后处理简单、很少污染环境、选择性高等特点,可在较高温度范围内使用,扩大了热力学上可能进行的酸催化反应的应用范围。 目录 介绍 物质资料 载体的改性 引入稀土元素 失活机理 表征技术 物质特性 优势 介绍 物质资料 载体的改性 引入稀土元素 失活机理 表征技术 物质特性 优势 研究意义 展开 介绍 因为环境污染问题,在环保呼声日益高涨、强调可持续发展 固体超强酸 的今天,已是到了非解决不可的地步。自20世纪40年代以来,人们就在不断地寻找可以代替液体酸的固体酸,固体超强酸更是成为热门研究对象。固体酸克服了液体酸的缺点,具有容易与液相反应体系分离、不腐蚀设备、后处理简单、很少污染环境、选择性高等特点,可在较高温度范围内使用,扩大了热力学上可能进行的酸催化反应的应用范围。 物质资料 固体超强酸 酸催化反应涉及到烃类裂解、重整、异构等石油炼制过程,还涉及到烯烃水合、烯烃聚合、芳烃烷基
化、芳烃酰基化、醇酸酯化等石油化工和精细化工过程,可以说酸催化剂是这一 固体超强酸 系列重要工业的基础。在这些生产过程当中应用的酸催化剂主要还是液体酸,虽然其工艺已很成熟,但在发展中却给人类环境带来了危害,同时也存在着均相催化本身不可避免且无法克服的缺点,如易腐蚀设备,难以连续生产,选择性差,产物与催化剂难分离等原因。 从而从液体含卤素超强酸发展为无卤素固体超强酸、单组分固体超强酸、多组分复合固体超强酸。无论是催化剂的制备、理论探索、结构表征,还是工业应用研究都有了新的发现,固体超强酸由于其特有的优点和广阔的工业应用前景,已受到国内外学者广泛关注,成为固体酸催化剂研究中的热点。人们在不断开发新的固体酸催化剂和固体酸催化工艺的同时,也在不断地探讨固体酸的酸性形成的机理,探讨固体酸催化反应的机理。本文重点对固体超强酸改性、理论研究、表征技术、失活机理及应用领域进行综述,并指出了固体超强酸催化剂今后研究和开发的主要方向。 载体的改性 催化剂 固体超强酸催化剂 在单组分固体超强酸催化剂的应用中,人们发现主要活性组分s一在反应中较易流 分子式 失,特别是在较高温度条件下容易失活,这类单组分固体催化剂虽然有较好的起始催化活性,但单程寿命较短。通过对催化剂载体的改性,使催化剂能提供合适的比表面积、增加酸中心密度、酸种类型、增加抗毒物随着人们对固体超强酸不断深入研究,催化剂能力、提高机械强度等作用。目前改性研究的方向主要有:以金属氧化物zK)2、Ti02和Fe2Ch为母体,加入其他金属或氧化物,形成多组元固体超强酸;引入稀土元素改性;引入特定的分子筛及纳米级金属氧化物等。 引入其他金属或金属氧化物 固体超强酸催化剂的制备对金属氧化物有特殊要求。有些氧化物如MgO、 固体超强酸
固体超强酸概述
固体超强酸概述 超强酸是比100%的H2S04还强的酸,其Ho<-11.93。许多重要的工业催化反应都属于酸催化反应,而固体酸和液体酸相比,具有活性和选择性高、无腐蚀性、无污染以及与催化反应产物易分离等特点,被广泛地用于石油炼制和有机合成工业。常用的固体酸催化剂有分子筛、离子交换树脂、层柱粘土等,它们的酸强度一般低于Ho= —12.0,对需要强酸的反应存在一定的局限性。20世纪60年代初,Olah等发现的HS03F-HF、HF-SbP5等液体魔酸,虽然其酸强度非常高,Ho高达—20.0以上,甚至甲烷在这种液体超强酸中都能质子化,但因其具有强腐蚀性和毒性,以及催化剂处理过程中会产生“三废’’等问题,难以在生产实际中应用。20世纪70年代初开始有人试图将液体超强酸如SbP5、HS03F-SbF5和HF-SbP5等负载到石墨、A1203和树脂等载体上,但仍不能解决催化剂分散、毒性和“三废’’等问题,未能工业应用。1979年Arata等首次报道了无卤素型SO42-/MxOy固体超强酸体系,发现某些用稀硫酸或硫酸盐浸渍的金属氧化物经高温焙烧,可形成酸强度高于100%硫酸104倍的固体超强酸。后来Arata等又将钨酸盐和钼酸盐浸渍Zr02制得WO3/Zr02、M003/Zr02固体超强酸,其酸强度虽比SO42-/Zr02稍低,但仍比100%硫酸高几百倍。1990年Hollstein等发现Fe、Mn和Zr的混合氧化物硫酸根制备的超强酸催化剂正丁烷异构化活性比SO42-/Zr02高1000倍以上。这类固体超强酸易于制备和保存,特别是它与液体超强酸和含卤素的固体超强酸相比,具有不腐蚀反应装置、不污染环境、可在高达500℃下使用等特点,引起人们的广泛重视。 固体超强酸主要有下列几类:(Ⅰ)负载型固体超强酸,主要是指把液体超强酸负载于金属氧化物等载体上的一类。如HF-SbF5-AIF3/固体多孔材料、sbP3-Pt/石墨、SbP3-HF /F-A1203、SbF5-FSO3H/石墨等。(Ⅱ)混合无机盐类,由无机盐复配而成的固体超强酸。如AICl3-CuCl2、MCl3-Ti2(SO4)3、A1C13-Fe2(S04)3等。(Ⅲ)氟代磺酸化离子交换树脂(Nation-H) (Ⅳ)硫酸根离子酸性金属氧化物SO42-/MxOy超强酸,如SO42-/Zr02、SO42-/Ti02、SO42-/Fe203等。(V)负载金属氧化物的固体超强酸,如W03/Zr02、M003/Zr02等。 在上述各类超强酸中,(Ⅰ)—(Ⅲ)类均含有卤素,在加工和处理中存在着“三废”污染等问题。(Ⅳ)、(V)类超强酸不含有卤原子,不会污染环境,可在高温下重复使用,制法简便。本节着重对这两类超强酸进行介绍。 MxOy型固体超强酸 (1)固体超强酸的制备 SO42-/MxOy型固体超强酸一般采用浓氨水中和金属盐溶液,得到无定形氢氧化物,然后再用稀硫酸或硫酸铵溶液浸渍、烘干和焙烧制得。然而,金属盐原料、沉淀剂、浸渍剂不同对制备的氧化物、超强酸的表面性质影响很大,制备环境如焙烧温度、沉淀温度、金属盐溶液浓度、pH、加料顺序、陈化时间及SO42-浸渍浓度也很重要。如何改善制备条件获得高质量、高酸性的固体超强酸是该类材料研究的最基本的问题。 (A) 金属氧化物的选择: