toll样受体作为模式识别受体在先天免疫中的作用

模式识别受体在先天免疫中的作用:Toll样受体最新研究进
展
Toll样受体作为识别病原保守结构的成分,其发现大大促进了我们对机体是如何感知病原入侵、引起先天免疫反应并启始针对特定病原的适应性免疫反应的认识。尽管TLRs对宿主防御是很关键的,但已经越来越明确,TLR信号传递负性调节的缺失,以及TLRs对宿主自身分子的识别,都与炎症反应和自身免疫疾病的发病机理有密切的关系。而且,现在已经清楚的知道,TLRs和最近证实的胞浆先天免疫感受器的相互作用,对发动有效的免疫反应是至关重要的。本文将阐述TLR在宿主防御和疾病中的生物学作用的最新研究进展。
在过去的十年中,人们对先天性免疫识别细菌成分以及它在宿主抵抗感染中的关键作用的认识,已经取得了极大的进步。早期的观点认为先天性免疫反应非特异性识别细菌;然而,在19世纪90年代中期,TLRs的发现表明:先天性免疫对病原的识别实际上是特异的,它依赖由生殖细胞编码的PRRs,PRRs已经进化为识别涉及病原相关分子模式(PRRs)的外源病原成分。TLRs是I型跨膜蛋白,包括富含亮氨酸重复区的胞外区、跨膜区和胞内区。胞外区介导PAMPs的识别;胞内区的TIR结构域是信号向下游转导所必需的。截至目前,已经分别在人和小鼠身上发现了10种和12种可以发挥功能的TLRs,其中的TLR1-TLR9
是二者共有的。由于一种逆转录病毒的插入,小鼠的TLR10并不发挥作用,而TLR11、TLR12和TLR13已经在人的基因组中丢失。对每一种TLR缺陷型小鼠的研究表明,它们各自在PAMPs识别和免疫反应中发挥不同的作用。几种TLR胞外区晶体结构的说明为证实一些PAMPs可以充当TLRs的配体提供了结构上的认识。TLRs可以识别来自于细菌、病毒、真菌和寄生虫等微生物的包括脂质、脂蛋白、蛋白和核酸等成分。PAMPs可在细胞的不同部位被TLRs识别,这些部位包括胞浆膜、内噬体、溶酶体和内噬溶酶体。TLRs在细胞上的合适定位,对于配体结合的难易、对诸如核酸之类的自身分子耐受的维持以及下游信号转导都是至关重要的。
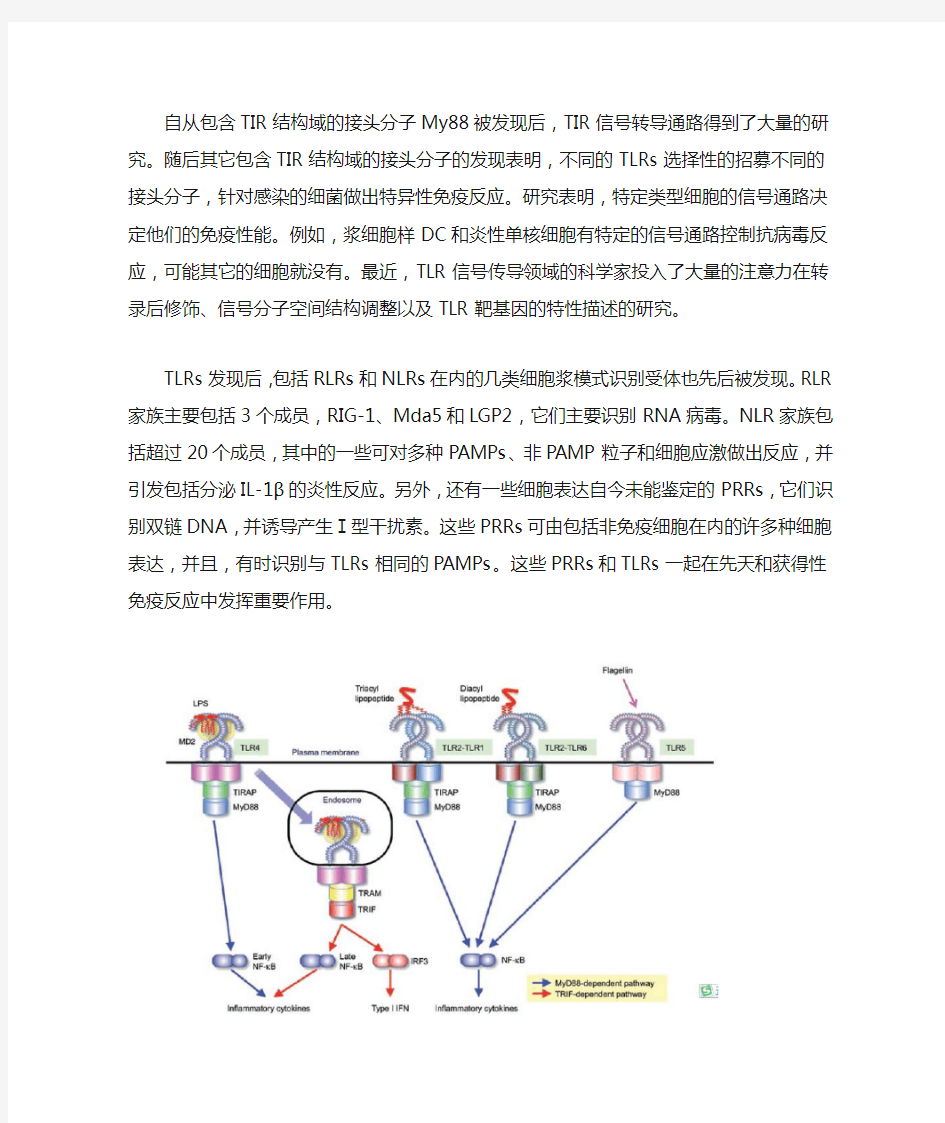
自从包含TIR结构域的接头分子My88被发现后,TIR信号转导通路得到了大量的研究。随后其它包含TIR结构域的接头分子的发现表明,不同的TLRs选择性的招募不同的接头分子,针对感染的细菌做出特异性免疫反应。研究表明,特定类型细胞的信号通路决定他们的免疫性能。例如,浆细胞样DC和炎性单核细胞有特定的信号通路控制抗病毒反应,可能其它的细胞就没有。最近,TLR信号传导领域的科学家投入了大量的注意力在转录后修饰、信号分子空间结构调整以及TLR靶基因的特性描述的研究。
TLRs发现后,包括RLRs和NLRs在内的几类细胞浆模式识别受体也先后被发现。RLR 家族主要包括3个成员,RIG-1、Mda5和LGP2,它们主要识别RNA病毒。NLR家族包括超过20个成员,其中的一些可对多种PAMPs、非PAMP粒子和细胞应激做出反应,并引发包括分泌IL-1β的炎性反应。另外,还有一些细胞表达自今未能鉴定的PRRs,它们识别双链DNA,并诱导产生Ⅰ型干扰素。这些PRRs可由包括非免疫细胞在内的许多种细胞表达,并且,有时识别与TLRs相同的PAMPs。这些PRRs和TLRs一起在先天和获得性免疫反应中发挥重要作用。
图Ⅰ:PAMP被细胞表面的TLRs识别,TLR4和MD2一起与LPS结合形成复合物。LPS六条链中的5条与MD2结合,剩下的一条与TLR4结合。受体多聚体的结构是由两个拷贝的TLR4-MD2-LPS复合物组成,通过招募包含TIR结构域的接头分子TIRAP(mal)和MyD88(MyD88依赖通路)来启动NF-κB早期活化的信号传递。继而,TLR4-MD2-LPS 复合体被内在化并保留在内噬体中,在内噬体中它通过招募TRAM和TRIF来引发导致IFR3和晚期NF-κB活化的信号转导,进而诱导Ⅰ型干扰素的生成(TRIF依赖通路)。早期和晚期NF-κB的活化对于炎性细胞因子的诱发都是必要的。TLR2-TLR1和TLR2-TLR6异二聚体分别识别三酰脂肽和二酰脂肽。三酰脂肽的三条链中的两条与TLR2结合,另一条与TLR-1(没有TLR-6)的疏水通道结合。TLR2-TLR1和TLR2-TLR6通过招募TIRAP和MyD88
来诱导NF-κB的活化。TLR5识别鞭毛蛋白,并通过MyD88活化NF-κB。
尽管TLRs是抗感染的必不可少的保护性免疫,但不合适的TLR反应会导致严重的慢性炎症反应和全身性自身免疫疾病。确实,TLR介导的反应的负反馈调节缺失的小鼠会患这些疾病。更重要的是,越来越多的证据表明,死亡细胞产生的内源分子或是某些病理状态会刺激TLRs,并导致炎性反应和自身免疫疾病的发展或加速。
在这篇文章中,我们研究了结构生物学、细胞生物学以及TLRs信号转导的现有认识,然后,我们会描述TLRs和胞浆PRRs在适应性免疫反应中的作用,最后,探讨TLR介导的内源分子的识别,以及它们在免疫系统中的作用的最新研究进展。
细胞表面TLRs的结构和配体
根据TLRs在细胞的结合位置和各自的PAMP配体,主要将其分成两个亚家族。一个由TLR21、TLR2、TLR24、TLR5、TLR6和TLR11组成,它们在细胞表面表达,并识别细菌表面的成分,如脂、脂蛋白和蛋白;另一家族有TLR3、TLR7、TLR8和TLR9构成,它们仅在胞内小泡中表达,如内质网、内噬体、溶酶体和内噬溶酶体,它们识别微生物核酸。
TLR4,作为TLR家族最早发现的成员之一,被确认正是长期探求的对LPS做出反应的受体,LPS是革兰氏阴性菌外膜的成分,可以引起脓毒性休克。TLR4与MD2在细胞表面形成复合物,它们共同充当LPS的主要结合构件。对TLR4-MD2和LPS形成的复合物的
结构的研究表明:LPS六条脂肪酸链中的五条结合在MD2的疏水袋上,而剩下的一条链被暴露在MD2的表面与TLR4结合。磷酸基团也与带正电的TLR4残基相互作用。受体多聚体的最终构造是由两个TLR4-MD2-LPS复合物组成的,它通过招募胞内接头分子来启动信号转导。其它的一些蛋白,如LBP和CD14等,也参与了LPS的结合。LBP是可结合LPS 的胞浆可溶蛋白,CD14是一个由糖基磷脂酰肌醇连接的,包含富含亮氨酸重复的蛋白,该蛋白可结合LBP,并传递LPS-LBP给TLR4-MD2复合物。除了结合LPS,TLR4还参与呼吸道合胞病毒融合蛋白、小鼠乳房肿瘤病毒封装蛋白、肺炎链球菌溶血素和植物源的抑制细胞生长的药物紫杉醇的识别,尽管TLR4和这些配体相互作用的在结构上的认识还没有得到证明。
TLR2广泛参与来自细菌、真菌、寄生虫和病毒的PAMPs的识别,包括细菌表面的脂肽、革兰氏阳性菌的肽聚糖和磷壁酸、分支杆菌的阿拉伯木聚糖脂、真菌的酵母聚糖、克氏锥虫的tGP2粘蛋白以及来自麻疹病毒的红血球凝集素蛋白。TLR2通常与TLR1或TLR6
形成异二聚体。TLR2-TLR1异二聚体特异识别革兰氏阴性菌的三酰脂肽和支原体,而
TLR2-TLR6异二聚体识别革兰氏阳性菌的二酰脂肽和支原体。有研究从结构认识上证实了这些异二聚体区分脂肽结构的机制。TLR2-TLR1和TLR2-TLR6共享一个m样结构(图1)异二聚体在TLR2-TLR1-配体复合物上,pam3CSK4的三条链中的两条与TLR2结合,而第三条链与TLR1的疏水通道结合。这样三酰脂肽的识别就实现了。然而,因为TLR6没有疏水通道,所以TLR2-TLR6异二聚体就不能够识别三酰脂肽。同时,TLR1和TLR6不同的脂质结合袋都负责脂蛋白的区分。而且,TLR2能够与细胞表面协助PAMP识别的共受体一同起作用。这些共受体包括CD 36和dectin-1,CD36可与TLR2-TLR6异二聚体共同介导部分TLR2激动剂的感知,而dectin-1是一种C型凝集素,可以结合真菌β葡聚糖,并诱导其内在化。尽管确信,TLR2激动剂主要诱导产生炎性细胞因子、巨噬细胞的非Ⅰ型干扰素及DCs,它还可以响应牛痘病毒感染并诱使炎性单核细胞产生Ⅰ型干扰素,这表明,TLR2在抗病毒反应中具有针对特定类型细胞的作用。看起来,通常是诱使Ⅰ型干扰素产生因素的核酸,并不参与TLR2的活化。
TLR5识别细菌鞭毛的鞭毛蛋白成分(图1)。小肠的CD11c+CD11b+固有层DCs对TLR5有高表达量。固有层DCs在促进产生IL-17的T H17细胞和T H1细胞的分化有非常特殊的作用,同时,对初始B细胞分化成产生IgA的浆细胞以应对鞭毛蛋白也有促进作用。而且,固有层DCs可以产生视黄酸,这使得体液免疫和细胞免疫都能够实现。小鼠的肾脏和膀胱相对于TLR5而言对TLR11有高表达量。TLR11被认为是用来识别导致肾盂肾炎细菌的成分的,因为TLR11缺失的小鼠容易感染这种细菌。TLR11还可以识别来自兔弓形虫的肌动蛋白样分子。
核酸感应TLRs的结构和配体
TLR3最初被认为是用来识别模拟合成的双链RNA(dsRNA)、聚肌胞苷酸(poly(I:C))的,它模拟病毒感染,并通过促进Ⅰ型干扰素和炎性细胞因子的产生来引发抗病毒免疫反应。识别的机制是通过对人的TLR3胞外区结合dsRNA的结构分析来阐明的(图2)。TLR3胞外区有一个大的马蹄样结构,它可能是为了增加表面积并帮助识别dsRNA.在TLR3胞外区凸出表面的侧面,dsRNA结合在N端和C端两个不同的位置,这为TLR3通过C端区域形成同二聚体提供了足够的稳定性。除了识别poly(I:C),TLR3还识别呼吸道肠道病毒的基因组RNA、单链RNA复制过程中产生的双链RNA、病毒(包括呼吸道合胞病毒、脑心肌炎病毒和西尼罗河病毒)以及某些小分子干扰RNA。TLR3通过产生Ⅰ型干扰素和炎性细胞因子来触发抗病毒免疫反应,这表明在抗病毒感染中发挥关键作用。TLR3缺陷的小鼠易于遭受小鼠巨细胞病毒的致命感染,而TLR3缺陷的人更易感染HSV-1。
TLR7最初被确认识别咪唑喹啉衍生物,如咪喹莫特和瑞喹莫德(R-848)和鸟嘌呤类
似物,如罗唑利宾(有抗病毒和抗肿瘤的特点),识别来自RNA病毒的单链RNA,如水泡性口膜炎病毒、A型流感病毒和人免疫缺陷综合症病毒(图2)。TLR7还识别合成的多聚尿嘧啶RNA和某些小分子干扰RNA。TLR7在pDCs有高表达量,pDCS在感染病毒后可以
产生大量的Ⅰ型干扰素,而pDCs应答RNA病毒诱导产生的细胞因子完全依赖TLR7,这表明TLR7充当单链RNA病毒的感受器。而且,cDCs上表达的TLR7感应细菌RNA类型包括B型链球菌并诱导产生Ⅰ型干扰素。pDCs的TLR7介导的RNA病毒的识别以复制独立
的方式发生。病毒被内在化并被招募到内噬溶酶体上,继而触发TLR7介导的对ssRNA的
识别并起始抗病毒反应。而且,TLR7还可以识别复制的水疱性口炎病毒,它通过自噬的方式进入胞浆,自噬是溶酶体降解胞内蛋白的过程,这涉及到称为自噬体的具有双层膜的小泡的形成(图2)。在感染水泡性口膜炎病毒后,缺失可诱导自噬体形成的自噬相关蛋白Atg5的pDCs表现出产生α型干扰素缺陷。pDCs表现出基本的自噬体形成。这些发现表明,pDC 对于递呈胞质病毒复制中间体给溶酶体是至关重要的,在溶酶体中,TLR7参与它们的识别和随后的抗病毒反应的起始。
TLR8与TLR7是在进化上最相似的。TLR8介导R-848和病毒ssRNA的识别。相对于TLR7缺陷的小鼠,TLR8缺陷的小鼠对这些激动剂反应正常。TLR8在许多组织中都有表达,而在单核细胞中表达量最高,细菌感染后还会上调表达量。
TLR9识别未甲基化的2′-脱氧(CpG)DNA基元,这一结构多见于细菌和病毒,而极少在哺乳动物细胞中出现(图2)。合成的CpG寡聚核苷酸充当TLR9的配体并直接活化DCs、巨噬细胞和B细胞,还促使强烈的T H1反应。DNA寡核苷酸有磷酸骨架时,DNA糖骨架
的2′脱氧核糖对于TLR9的识别是非常重要的。相反,在非天然的硫代磷酸骨架存在时,CpG基元是必不可少。TLR9在pDCs中有高表达量,而且它充当DNA病毒感染(如,小
鼠巨细胞病毒、HSV-1和HSV-2)的感受器。除了识别DNA,TLR9还直接识别不溶性的结晶疟原虫色素,结晶疟原虫色素是作为恶性疟原虫消化宿主血红蛋白后排毒过程的副产品而产生的。
核酸感应TLRs的细胞定位
如前所述,核酸感应TLRs固定在细胞内的许多个不同的部位。内噬溶酶体酸化过程的阻断能够抑制TLR7和TLR9诱导的反应,这一发现表明,内在化的核酸分子被递呈给内噬溶酶体对于这些TLRs的相互作用是至关重要的。在未受刺激的细胞中,TLR9和TLR7被
专门的隔离在内质网中,当接受配体刺激后,迅速的传递给内噬溶酶体(图2)。这种转运
是由定植在内质网上的蛋白UNC93B1来调节的,UNC93B1是一个跨12层膜的蛋白。带有一个编码UNC93B1基因错义突变的小鼠,在应答TLR7和TLR9配体以及TLR3配体时,有产生细胞因子和上调共刺激分子的缺陷,这种小鼠对细菌和病毒高度易感。据报道,UNC93B1缺陷与人类患者的HSV-1脑炎有关。来自这些患者的细胞对TLR3、TLR7和TLR9激动剂有低反应性,但是对细胞外的TLRs有完整的反应。UNC93B1特异结合内质网中TLR3、TLR7和TLR9的跨膜区,而且TLR7和TLR9在带有3d变异的DCs中不离开内质网。这
些结果共同表明,UNC93B1协助TLR7和TLR9从内质网到内噬溶酶体的传递,这对于这
些TLRs诱发免疫反应是必要的。TLRs的运输还受到另两个保留在内质网中的蛋白PRAT4A 和gp96的调控。PRAT4A分别连接TLR4和TLR9,对于TLR4和TLR9分别被转运到胞
浆膜和内噬溶酶体都是必要的。PRAT4A缺陷的细胞没有对TLR2、TLR4和TLR9激动剂
的反应,而TLR3介导的反应在这些细胞中是完整的,这表明TLR3和TLR9转运的调控是不同的。
TLR9是已知的有内噬溶酶体中的胞内蛋白酶降解的,它产生一个介导配体识别并起始
信号转导的功能受体(图2)。可能介导TLR9降解的蛋白酶包括组织蛋白酶,如组织蛋白
酶B,组织蛋白酶S,组织蛋白酶L,组织蛋白酶H和组织蛋白酶K,以及天冬氨酸肽链内切酶。然而,关于TLR9功能的降解依然存在争议。N末端区域的特殊富含亮氨酸重复的降解使得TLR9不能对配体反应,并且TLR9带正电的N末端区域可能介导与CpG DNA的结合,这表示完整结构在TLR9活化中的重要作用。
图2:胞内TLRs识别PAMP。TLR3识别来自病毒或被病毒感染的细胞的dsRNA。dsRNA 结合在TLR3胞外区凸出表面的侧面的N端和C端两个位置,这使得TLR3通过C端区域形成同二聚体。TLR3活化TRIF依赖通路,诱导产生Ⅰ型干扰素和炎性细胞因子。在pDCs,TLR7识别来自内噬溶酶体的ssRNA病毒的ssRNA,并通过MyD88分别活化NF-κB和IRF7,并诱导产生炎性细胞因子和Ⅰ型干扰素。另外,自噬体也参与了递呈ssRNA到表达TLR7的小泡的过程。TLR9识别来自细菌和病毒的RNA。TLR9通过胞内蛋白酶溶蛋白性降解对向下游的信号传导是必要的。在pDCs中TLR9通过招募MyD88来活化NF-κB和IRF7。TLR3, TLR7 和TLR9主要以稳定结合的形式定植在内质网上,并转移到内噬溶酶体,然后结合他们的配体。在内质网中,UNC93B1与这些TLRs相互作用,并且对这种转移是必不可少的。gp96缺陷的巨噬细胞在应对TLR1, TLR2, TLR4, TLR5, TLR7和TLR9受体激动剂时,缺乏细胞因子诱导反应,gp96是内质网热休克蛋白90家族的成员。而且已证实gp96结合TLR9.这些发现表明,不像只调控某些特定TLRs转移的PRAT4,gp96担当TLRs的通用伴侣蛋白。
TLR信号传导中包含TIR结构域的接头分子
每个TLR都引发特定的生物学反应。如,TLR3和TLR4都产生Ⅰ型干扰素和炎性细胞因子反应,而细胞表面TLR1-TLR2,TLR2-TLR6和TLR5主要诱导炎性细胞因子(图1和2)。这些不同由于包含TIR结构域的接头分子的发现而得到解释,这些接头分子包括MyD88, TIRAP(Mal),TRIF和TRAM,它们被各自的TLR招募,并活化各自的信号通路(图1和2)。MyD88是第一个被发现的TIR家族成员,被除了TLR3外的所有TLRs使用,并活化转录因子NF-κB和有丝分裂原蛋白激酶MAPKs,进而诱导炎性细胞因子。相反,TRIF被TLR3和TLR4使用,并诱导其它的通路,进而活化转录因子IRF3和NF-κB,随后诱导Ⅰ型干扰素和炎性细胞因子。TRAM 和TIRAP充当类型接头分子,它分别为TLR4招募TRIF,为TLR2和TLR4招募MyD88。因此,TLR信号通路可以大致的分为MyD88依赖型通路和TRIF 依赖型通路,MyD88依赖型通路驱动诱导炎性细胞因子,而TRIF依赖型通路同时负责Ⅰ型干扰素和炎性细胞因子的诱导。
TLR4是唯一的同时利用四个接头分子并活化两个通路的TLR(图1)。TLR4最初招募胞浆膜的TIRAP,随后帮助招募MyD88来触发NF-κB和MAPKs的初始活化。TLR4随后经历动力依赖的内吞作用,并被转移给内噬体,然后形成一个有TRAM和TRIF而不是TIRAP和MyD88的信号复合体,进而引发TRIF依赖通路的活化,导致IRF3的活化以及晚期NF-κB和MAPK的活化。因此,TLR4活化MyD88依赖型通路早于TRIF依赖型通路。特别地,两个通路的活化对通过TLR4信号传导的炎性细胞因子的诱导都是必要的,与此相反,对于其它的TLRs,任何一个信号通路的活化都足以诱导产生炎性细胞因子。为什么单一信号通路的活化不能满足通过TLR4信号通路诱导产生的炎性细胞因子需要,仍然是个谜。
MyD88依赖通路
TLRs被它们同源的PAMPs结合后,MyD88招募IL-1相关受体激酶IRAK4, IRAK1, IRAK2 和IRAK-3(图3)。IRAK4最先被活化,并对NF-κB和MAPK下游的MyD88的活化发挥关键作用。IRAK的活化导致与TRAF6的相互作用,一个E3连接酶催化连接到靶蛋白上Lys63(K63)的多聚泛蛋白的合成,包括TRAF6自身和IRAK1协同二聚E2泛素结合酶UBC13和Uev1A。连接有K63的多聚泛蛋白链结合到TAB2和TAB3的新型锌指型泛蛋白结合域来活化TAK1,TAB2和TAB3是激酶TAK1复合体的调节成分。连接有K63的多聚泛蛋白链还结合到NEMO的泛蛋白结合结构域,NEMO是IKK复合体的必要调节成分,NF-κB的活化需要它。因此,K63多聚泛蛋白链可能负责招募TAK1来形成IKK复合体,然后允许TAK1通过它临近IKK复合体的部分使IKKβ,通过磷酸化和随后的ⅠκB蛋白的降解使NF-κB活化。然而,Ubc13缺陷的细胞对TLR激动剂表现出正常的NF-κB活化,不管NEMO的连接有K63的多聚泛蛋白是否缺陷,这表明在NEMO介导的NF-κB活化中存在不依赖连接K63的泛蛋白的机制。由线状泛素链组装复合的NEMO的头尾相连的线性多聚泛素化已被证明在IKK活化过程中是非常重要的。
TAK1通过诱导MAPK激酶的磷酸化(而不是泛素化)同时活化MAPKs Erk1, Erk2, p38和Jnk,然后活化多种转录因子,诱导AP-1,同时影响翻译。不管有没有正常的NF-κB活化,Ubc3缺陷型细胞表现出削弱的MAPK的活化。但是,负责MAPK活化的Ubc3的直接靶标仍是未知。
MyD88通路的活化导致许多基因的启动,而且其中的一些对依赖NF-κB的转录有关键作用(图3)。这些基因包括I κ B蛋白I κ B δ,它充当NF-κB p50亚基的共诱导活化者,帮助IL-6和IL-12p40的诱导产生;C/EBP δ,它和NF-κB一同最大化IL-6的产生;I κ B-NS,它通过调节NF-κ B p65亚基的DNA结合活性抑制IL-6和TNF;以及ATF3,它通过招募组
蛋白脱乙酰基酶来限制NF-κB活化。
TRIF依赖的通路
TRIF依赖的通路在IRF3和NF-κB都活化后达到高峰(图3)。TRIF招募TRAF6并活化TAK1用于NF-κB的活化,这一过程有可能是通过依赖泛化素的机制,这一机制与依赖MyD88的途径相似。TRIF也可以通过同型RIP相互作用来获得RIP1. TLR3会活化激活与K63有关连的泛素,RIP1会经过这一泛素的处理,并且这一处理过程需要NF-κB的激活。TRADD伴随着RIP1,缺少TRADD的细胞RIP1泛化素有缺陷,并且得不到NF-κB的活化,这就意味着TRADD参与的RIP1的激活发生在TLP3的下游。Pellino-1是环状Pellino家族的一份子,其控制包括E3的泛素连接,Pellino-1缺失会导致RIP1泛化素的缺失,并且NF-κB无法激活,以响应TLR3受体激动素的作用,尽管MyD88途径的NF-κB激活仍然正常。综上所述,TRIF与TRAF6, TRADD, Pellino-1 ,RIP1组成高蛋白分子传递复杂的信息以用于TAK1的激活,TAK1的激活又反作用于NF-κ B 与MAPK的激活。
除了能引导NF-κB的激活以外,TRIF的依赖途径还能引导IRF3的激活与β干扰素的转录。TRIF招募一个包括IKKs TBK1 和IKKi (IKKε)在内的信息传递复合体,这个复合体可以促进IRF3的磷酸化,并且可以引导其向细胞核方向的转运。TBK1-IKKi被TRIF激活需要TRAF3. TRAF3的缺失会损害TLR3引导β干扰素形成的过程,同时对于TLR7, TLR9 和RLR的引导过程也有影响,这些都表明TRAF3在不同核酸的PRR的β干扰素引导中起着十分重要的作用。
TLR4的信号传递过程中TRAF3也会合并入MyD88的复合体上。但是,是将TRAF3依附于K48关联的泛素化过程,之后经cIAP1 与cIAP2降解。cIAP1 与cIAP2都是MyD88信号转导复合物的一部分,但不属于TRAF3复合物。TRAF3的促使降解膜近端的信号转导复合物的转移至细胞质这一过程又促使了TAK1的激活。这些发现都表明TRAF3能促进IRF3的激活,同时抑制MyD88依赖途径。依赖MyD88与TRAF3的依赖途径的在调控上的不同,是通过单一分子,这已在NRDP1的研究中有报告。NRDP1是一个包含环状E3连接酶,其可与TBK1相互作用并且可以通过K63关联的泛化素加强TBK1的激活过程,对上述过程,需要Ubc13的参与。同时它通过它的相互作用,以及MyD88的降解来抑制依赖MyD88的途径。通过这些分子来平衡应激细胞因子和Ⅰ型干扰素的产生量,可能在控制肿瘤细胞扩散和自身免疫疾病起至关重要的作用。
pDCs中的TLR7和TLR9信号通路
TLR7和TLR9信号通路已经得到了广泛研究,试图解释病毒感染后它们诱导产生Ⅰ型干扰素的可能性。pDCs中TLR7和TLR9的特殊之处在于它们都需要MyD88来诱导Ⅰ型干扰素(图3)。在这种情况下,pDCs稳定表达的IRF7 结合MyD88并与IRAK4, TRAF6, TRAF3, IRAK1 和IKKα形成复合蛋白信号复合物,IRF7被IRAK1与或或IKK α磷酸化,从复合物上脱离,并转移到核内。除了需要磷酸化,IRF7活化可能还需要TRAF6和Ubc13依赖的泛素化。然而,IRAK1, IKK α 和TRAF3分别地参与了IRF7, MyD88, IRAK4的活化,而TRAF6对IRF7 和NF- κB的活化是关键的。
控制pDCs产生Ⅰ型干扰素的其它成分也已经被发现(图3)。OPNi是由TLR9诱导的一种骨架蛋白前体,它在胞浆中是沉默的,而在pDCs中是MyD88-IRF7复合体的成分。药物抑制磷酸肌醇3 - OH激酶(PI(3)K)能消除IRF7的核易位。而且,mTOR 和p70S6K 都是PI(3)K的下游靶标,mTOR 和p70S6K被抑制会打断TLR9和MyD88的相互作用,进而导致IRF7的核转移和Ⅰ型干扰素的诱导被削弱。这些发现表明,在病毒感染期间,
PI(3)K-mTOR通路加速pDCs产生Ⅰ型干扰素。IRF5被结合到MyD88复合物,并控制IL-6 和IL-12p40的诱导。MyD88-IRF5通路也被许多其它类型细胞的TLRs利用,如巨噬细胞和cDCs。敲除IRF8的pDCs表现出TLR9介导的Ⅰ型干扰素和炎性细胞因子诱导缺失,只有很少的NF- κB活化,这表明可能IRF8和NF- κB共同诱导细胞因子。
CpG-TLR9信号复合体在内涵体中的保留也是pDCs控制抗病毒先天免疫反应的重要机制。A(D)型CpG寡脱氧核苷酸包含一个单一的CpG基元和一个多聚(G)尾巴在硫代磷酸二脂键骨架上,它可以诱导pDCs分泌Ⅰ型干扰素。它和TLR9, MyD88和IRF7一同pDCs 的早期内涵体中稳定保留。相反,B(k)型CpG寡脱氧核苷酸包含多个CpG基元在磷酸骨架上,它可以诱导cDcs产生IL-12和B细胞的活化,它迅速转移到晚期内涵体和溶酶体,导致很少的IRF7活化。
图3:TLR信号通路的概述。TLR介导的反应主要有MyD88依赖型通路和TRIF依赖型通路控制,MyD88依赖型通路被除了TLR3外的所有TLR使用,而TRIF依赖型通路为TLR3和TLR4使用。TRAM 和TIRAP分别是TLR4和TLR2-TLR4复合体的类型接头分子。在cDCs和巨噬细胞,MyD88招募IRAK4, IRAK1, IRAK2 和TRAF6,并通过活化NF- κ B, MAPK 和IRF5来诱导炎性反应。TRAF6与TAB2和TAB3一同活化TAK1,并活化IKK 复合体构成NeMO 和IKKαβ,并催化IκB蛋白用于磷酸化。NF-κ B 诱导C/eBPδ , I κBδ, I κB-NS, Zc3h12a, ATF3 和tristeraprolin (TTP),进而影响基因编码IL-6, IL-12p40 或TNF。TRIF 招募TRAF6, TRADD 和TRAF3。TRADD 和Pellino-1 and RIP1相互作用。RIP1 和TRAF6 共同活化TAK1,导致MAPK 和NF- κ B的活化。TRAF3 活化激酶TBK1 和IKKi 进而磷酸化并活化IRF3,最后,控制Ⅰ型干扰素的转录。Nrdp1参与TBK1-IKKi 的活化。TLR4信号传递过程中,TRIF依赖通路导致炎性激活。在pDCs, TLR7 和TLR9与IRAK4 和TRAF6一起招募MyD88,活化诱导炎性细胞因子的IRF5 和NF- κ B,还活化诱导Ⅰ型干扰素的IRF7。对于IRF7活化,需要IRAK1- 和IKK α –依赖的磷酸化,而且TRAF3位于这些激酶的上游。OPNi 参与IRF7 活化,并且OPNi参与IRF7 活化, IRF8促进NF- κB 激活。PI(3)K-mTOR-p70S6K 轴加强TLR7 和TLR9信号通路。IRF1在cDCs 而不是
pDCs中通过TLR7 and TLR9参与I 型干扰素的诱导。在已发现的TLRs负调节因子中,TANK (抑制TRAF6), A20 (抑制TRAF6和RIP1), A TG16A (抑制炎性激活) and SHP-1 (抑制IRAK1 和IRAK2)被认为是阻止由于增强的或延长的TLRs信号引起的炎性疾病。黄色,TLRs;绿色,刺激因子; 粉色, 负调节因子; 蓝色,靶基因。
TLR信号的负调节
TLR诱导反应的负调节,对于抑制炎性反应和有害的免疫反应是非常重要的。目前,已经发现了在多个水平上抑制TLR信号通路的负调节因子。这些负调节因子包括接头分子的剪切变异体及其相关蛋白,泛素连接酶,脱泛素酶,转录调节因子和小RNA。本文我们重点关注了TLR介导免疫反应的负调节因子,其破坏或变异导致体内持续的炎症。
TANK 结合TBK1和IKKi,并且与NF- κB 和IRF3的活化都有联系。TANK缺陷的小鼠自发形成自身免疫性肾小球肾炎,这种情况可以通过用抗菌素治疗抑制,或是MyD88和IL-6缺乏。尽管有完整的Ⅰ型干扰素诱导反应,TANK缺陷的巨噬细胞和B细胞表现出更多的NF- κ B活化和在应对TLR配体时产生更多的IL-6。TANK缺陷的细胞还表现出加强的TRAF6 泛素化。因此,TANK还充当巨噬细胞和B细胞TRAF6泛素化的负调节因子(图3)。
编码自噬相关分子Atg16L1的基因的变异被认为与克罗恩氏病有关。Atg16L1缺陷的小鼠高度易感葡聚糖硫酸酯钠引起的急性结肠炎,可以通过用IL-1 β和IL-18的抗体治疗来阻止。而且,来自这些小鼠的巨噬细胞表现出更多的半胱天冬酶-1的活化,响应LPS也产生更多的IL-1 β和IL-18。在缺少Atg16L1时,由LPS导致的半胱天冬酶-1过度活化需要TRIF,这表明Atg16L1负调节TRIF依赖通路,并导致半胱天冬酶-1活化(图3)。而且,取自Atg16L1缺陷小鼠的小肠潘氏细胞表现出应对小肠损伤相关基因的更高表达。因此,Atg16L1对小肠炎症抑制是必要的。
TLR刺激迅速诱导产生调节蛋白Zc3h12a,Zc3h12a包含一个CCCH型锌指结构域和RNA酶结构域。Zc3h12a通过RNA酶活性降解3′非翻译区的IL-6mRNA和IL-12p40mRNA。在应对TLR激动剂时,Zc3h12a缺陷的巨噬细胞持续产生显著多的IL-6和IL-12p40,但是正常量的TNF,而Zc3h12a缺陷的小鼠具有更高的血液免疫球蛋白水平和自身抗体产物。这些结果共同表明,Zc3h12a通过影响mRNA稳定性和阻止自身免疫来负调节TLR诱导的炎性反应(图3)。另一个锌指蛋白,Xfp36,可阻止自身免疫性关节炎的发生。Xfp36结合TNFmRNA3′非翻译区富含AU成分,并通过脱腺苷化移走poly(A)尾巴,进而导致降解(图3)。因此,这些锌指蛋白通过不同的机制来控制mRNA的稳定性。
A20是有TLR刺激诱导产生具有两种酶活性的蛋白,它可作为E3泛素连接酶和脱泛素化酶。体外分析表明,A20通过调节RIP1和TRAF6来限制NF- κ B的活化。A20缺陷型小鼠由于自发的多器官炎症和多种恶病质而过早死亡,这表明A20在体内有抗炎性作用。同时缺失A20和MyD88使小鼠免于过早死亡,并减少这些小鼠的炎症反应。而且,服用抗生素可防止恶病质。因此,A20可以禁止由共生菌诱发的TLR信号。带有编码酪氨酸磷酸化基因SHP-1变异的小鼠,形成炎性损伤与应答TLR刺激时巨噬细胞的异常活化有关。MyD88缺陷抑制这种炎症,这表明SHP-1负性调节MyD88依赖通路。据报道,SHP-1还抑制IRAK1 和IRAK2的功能。
PAMP识别中的非TLR胞浆PRRs
先天免疫系统通过RLRs、NLRs和一种未知的dsDNA感受器识别胞浆PAMPs。RLRs (RIG-I, Mda5和LGP2)是解旋酶,通过接头分子IPS-1来识别病毒的RNA和信号,诱导抗病毒反应。NLRs代表了PRRs的一个大家族,应答包括多种PAMPs、非PAMP粒子和细胞
应激在内的多种刺激。NLRs中,Nod1和Nod2识别细菌细胞壁成分的降解产物,而NLRP3 (NALP3)响应各种刺激形成炎性体复合物,通过半胱天冬酶-1促进IL-1β和IL-18的分泌。而且,细胞可通过未知通路触发Ⅰ型干扰素的诱导来响应来自病原的dsDNA和自身未能完全降解的dsDNA,尽管STING 和TBK1都是dsDNA触发信号所必需的成分。DAI (ZBP1-DLM1)已被确定为dsDNA的公认胞浆感受器,它在响应dsDNA时增加Ⅰ型干扰素的产生。但是,DAI缺陷型小鼠在用dsDNA刺激时有完整的Ⅰ型干扰素诱导,这表明DAI 是多余的。AIM2包含一个pyrin和HIN-200DNA结合结构域,与dsDNA结合,并与ASC 形成炎性体,触发IL-1 β产生。半胱天冬酶-1依赖的IL-1 β产生需要AIM2,但是,在应答dsDNA刺激,可递呈DNA给宿主细胞质的牛痘病毒(双链DNA病毒)和兼性细胞内的革兰阴性菌图拉弗朗西斯感染时,AIM2对于Ⅰ型干扰素的诱导不是必要的。而且,AIM2缺陷的小鼠相对于对照小鼠更易遭到F. tularensis的致命感染,可能是因为IL-1 β产生的缺陷。因此,AIM2是dsDNA诱导的IL-1 β生产而不是Ⅰ型干扰素的关键成分。磷酸酶Eya被认为是与STING和IPS-1相互作用的分子,可加强dsDNA和RLR信号传递中编码β干扰素的基因的启动子,尽管其生理学作用尚不清楚。这些类型的胞浆PRRs可由许多细胞表达,包括免疫和非免疫细胞,如成纤维细胞和上皮细胞,而且可识别与TLRs共有的PAMPs。在感染期间的获得性免疫反应的背景下,这些PRRS特有的作用和共有的作用都已经得到了研究。
在所有的PRRs中,TLRs主要在抗原递呈细胞如DCs和巨噬细胞,已经B细胞中表达,而且,许多TLR激动剂触发抗体反应以及T H1和T H17反应。多个水平的证据都表明TLR 在获得性免疫中有关键作用。已经被pDCs的TLR7和其它细胞的RIG-1感应的一个感染流感病毒的体内模型,表明发动B细胞和CD4+T细胞反应需要TLR7而不是RIG-1。在疫苗模型中,TLR7表现为保护性免疫。而且,感染淋巴细胞性脉络丛脑膜炎病毒后CD8+T细胞的分化需要TLR7,而不是RIG-1。这些数据共同证明了TLR7在诱导有效的抗病毒的获得性免疫反应中的作用。然而,TLRs还不足以诱导足够强烈的获得性免疫反应,因为CD8+T 细胞应答A型流感病毒时不需要TLR7或RLRs。A型流感病毒可以活化NLRP3f炎性体,触发IL-1 β产生,并且获得性免疫的形成也需要这一通路。尽管,有几种机制曾被认为是触发NLRP3炎性体活化的,但已证实,A型流感病毒诱导的NLRP3炎性体活化是独有的,病毒编码的M2离子通道负责触发这种活化。
虽然胞浆PRRs对形成适应性免疫的相对贡献仍不清楚,但它已被认为,非抗原呈递细胞(非- APC)的PRRS的胞浆信号参与促进了DC-介导的获得性免疫反应。Poly(I:C)被用作免疫佐剂,并由TLR3和Mda5识别。TLR3 在CD8 α+ DCs中有高表达量,CD8 α+ DCs 对凋亡的病毒感染细胞有高吞噬活性。TLR3缺陷型DCs在它们吞噬负载dsRNA的细胞和病毒感染的细胞时,不能够发动CD8+T细胞反应。因此,在DCs中TLR3识别从病毒感染细胞传递来的dsRNA触发DC变异,并将病毒抗原呈递给MHCⅠ类分子,这刺激CD8+T 细胞反应(图2)。这一过程,涉及到交叉引导,可以有最初被感染的细胞分泌的Ⅰ型干扰素和其它的细胞因子促进,在这些细胞中Mda5参与了病毒的识别和细胞因子的产生。实际上,一个用于评价poly(I:C)在TLR3和/或Mda5信号成分缺陷的小鼠中的佐剂效应的研究表明,针对特异抗原的抗体的产生和CD4+和CD8+T细胞的分化是同时受到TLR3和Mda5介导的通路的调控。而且,一个研究展示了在DCs、单核细胞和间质细胞中poly(I:C)被Mda5识别和结果诱导Ⅰ型干扰素的产生,为人类免疫缺陷病毒Gag蛋白疫苗设计的的小鼠模型的T H1反应需要这一过程。这表明,APCs和非APCs的PRR信号以及它们的相互作用足够发动一个强烈的获得性免疫反应。非抗原递呈细胞在构建获得性免疫反应中关键作用,已经由DNA疫苗的研究得到解释,DNA疫苗综合了有抗原序列的质粒和其它使先天免疫反应成为可能的因素。理想的B细胞和T细胞反应的诱导不需要TLR9、RLR或DAI,但
需要TBK1和STING。特别地,细胞转移实验已经解释了在造血细胞和非造血细胞中dsDNA 的识别都是DNA疫苗在体内的辅助性所需要的。
图4:TLRs和免疫疾病的内源激动剂。由死亡细胞如HMGB1释放的内源分子和热休克蛋白及ECM的成分被TLR2, TLR4或TLR2-TLR4识别。β淀粉和氧化的LDL都是被TLR4-TLR6与其共受体CD36一同识别。感染后产生的氧化的磷脂和抗菌肽β防御素2被TLR4识别。这些内源分子被细胞表面TLRs识别并导致炎症和修复反应。自身RNA和DNA 月LL37一同被内在化到内涵体,并分别被TLR7和TLR9识别。HMGB1自身的DNA复合物通过RAGE被内在化并被TLR9识别。包含免疫复合物的自身核酸分子同Fc受体如Fcγ RIIa被内在化,并刺激TLR7和TlR9。凋亡过程中自身DNA分子的不完全降解可能被胞内DNA感受器感知并活化TBK1。TLR7、TLR9和一种目前未知的DNA感受器识别自身核酸分子,导致Ⅰ型干扰素的产生,并促进自身免疫和炎性疾病。
细胞表面TLRs的内源配体
已经越来越明了,除了应答PAMPs,TLRs响应内源宿主分子并触发炎性反应。这些内源分子中的大多数都是由与细胞死亡、伤害和肿瘤细胞产生的,包括胞外基质的降解产物、热休克蛋白和高迁移率蛋白,它们充当细胞表面TLRs才刺激因子(图4)。而且,死亡细胞和含免疫复合物的自身抗原都含有自身核苷酸,由它们释放的染色体DNA和核蛋白复合物可刺激胞内TLR7和TLR9,并导致系统性自身免疫病的形成(图4)。
作为损伤和炎症的结果,ECM被胞内蛋白酶分解,并清除出细胞。据报道,部分被清除出细胞的成分可以活化TLR2或TLR4,或活化两者(图4)。这些成分包括双糖链蛋白聚糖、透明质酸、多功能蛋白聚糖、纤连蛋白额外域A和表面活性蛋白A。双糖链蛋白聚糖因诱导产生炎性细胞因子和趋化因子,并且这种诱导会因TLR2和TLR4 的缺陷而完全丧失。双糖链蛋白聚糖缺陷的小鼠对酵母聚糖和LPS导致的休克有高抵抗性,并具有低TNF凝聚
物和单核细胞渗透入肺,这这证明了biglycan-TLR2-TLR4通路在增强细菌诱导的肺损伤中的作用。透明质酸碎片在肺损伤后释放并聚积,能够刺激巨噬细胞通过TLR2和TLR4产生趋化因子。在透明质酸参与的非传染的肺损伤小鼠模型中,TLR2和TLR4双重缺陷的小鼠呈现低存活率,与较少的招募炎性细胞,增强的上皮细胞凋亡和更多的组织损伤,表明TLR2-TLR4介导的透明质酸的识别促进炎症和修复反应。额外区A和表面活性蛋白A也是有TLR4识别。
除了ECM成分,其它的细胞成分如HMGB1和热休克蛋白充当TLRs的配体(图4)。HMGB1是一种核非组蛋白,是脓毒性休克和缺血性再灌注模型的促炎性调节物,并被TLR2、TLR4或TLR9识别。HMGB1的无效化抗体抑制肝脏缺血再灌注模型的损伤,并且在这一模型中,TLR4缺陷的小鼠表现较低的损伤。这一发现表明,在非感染的状态下,TLR4响应内源分子并介导炎性反应。胞浆HMGB蛋白HMGB1, HMGB2和HMGB3作为通用的核酸感受器,活化TLRs、RLRs和DNA感受器的作用已被阐明。热休克蛋白,包括Hsp60, Hsp70, Hsp22 和gp96,也被认为与通过TLR2, TLR4或TLR2-TLR4活化巨噬细胞和DC,诱导促炎性调节物有关,尽管,尚不清楚这种效果是否是由于这些用大肠杆菌产物准备重组蛋白受到污染。
TLRs还参与某些病理状态下的炎性反应。在非感染的炎性疾病如动脉粥样硬化和阿尔茨海默氏病,氧化低密度脂蛋白蛋白和β -淀粉样蛋白,分别触发无菌炎症并以依赖于TLR4和TLR6的方式识别(图4)。这一识别可能是通过TLR4-TLR6异二聚体在清道夫受体CD36的协助下完成的。多功能蛋白聚糖是一个在肿瘤细胞中积聚的蛋白聚糖,通过TLR2、TLR6和CD14刺激肿瘤浸润髓样细胞产生TNF,加速肿瘤细胞新陈代谢。TLR2和多功能蛋白聚糖有直接联系。因此,TLR2对多功能蛋白聚糖的识别和最终的炎性环境可以支持肿瘤细胞的生存。
感染同样是TLRs识别的内源分子释放的触发因素(图4)。抗菌肽β防御素2,可直接中和入侵的微生物,它是由应答感染的黏膜组织和皮肤产生的,通过TLR4活化未成熟的DCS诱导上调共刺激分子,导致有效的免疫反应的诱导。TLR4和TRIF缺陷的小鼠可避免由于供给灭火的禽流感病毒引起急性肺损伤。这种病毒触发氧化磷脂的产生,氧化磷脂的产生承担通过TLR4-TRIF轴的急性肺损伤。因此,通过TLR4通路响应氧化应激是控制急性肺损伤的关键。
不合理的TLR介导的对自身核酸的识别
正常情况下,自身的核酸分子并不活化先天免疫反应。自身核酸分子在被内噬溶酶体中的TLRs识别前就已经被血清核酸酶合适的降解。TLR7和TLR9在细胞内的位置对于避免与胞外自身核酸分子接触是非常重要的。另外,蛋白水解成熟TLR9的(及可能TLR7 )用于防止泄漏到细胞表面的TLR9不合适的识别自身DNA是很重要的。可能这些保护在炎性和自身免疫状态下已经被破坏。例如,当自身核酸分子与内涵体蛋白形成复合物时,它们可能会变得抵抗核酸酶并增加与体内TLRs,反过来促进并维持自身免疫过程。而且,发现TLR7和TLR9从内质网到内噬溶酶体的转移是由LPS配体外的部分诱导的,这证明了炎性实际上可以增加核酸分子与TLR7和TLR9的接触的可能性。
在系统性红斑狼疮中,抗体与核酸或核蛋白有高凝聚。来自病人的血浆可促进pDCs产生Ⅰ型干扰素,且其浓度与病情严重程度有关。自身核酸分子或核蛋白被抗体结合后通过DCsd的Fc γ RIIa受体被内在化,然后传递给包含TLR7和TLR9的小泡,导致Ⅰ型干扰素的产生(图4)。而且,这些免疫复合物结合到B细胞表面抗原受体并被内在化以活化这些TLRs,导致自体反应B细胞的活化。因此,DCs和B细胞的协同活化,对于自身免疫疾病的形成和维持是很重要的。
HMGB1既可以结合抗原又可以结合自身DNA。这些HMGB1-DNA复合体结合到RAGE 上,并被转移到早期内涵体让TLR9识别,导致pDCs和B细胞的活化(图4)。LL37是由嗜中性粒细胞和角质形成细胞产生的抗菌肽,牛皮癣的皮肤损伤部位高表达LL37。LL37与坏死细胞释放的自身DNA和RNA形成聚合物,被内吞并保留在pDCs的内涵体中,然后分别活化TLR9和TLR7。在小鼠中发现的TLR7的复制体对TLR7配体有超反应。这些小鼠产生针对包含RNA自身抗原的抗体的同时还形成肾炎。降植烷是一个在植物中发现的异戊二烯烷烃,被注入到腹膜腔里后可诱导小鼠产生狼疮样疾病。随后的自身抗体的产生需要干扰素信号。这种情况下Ⅰ型干扰素产生的来源是未成熟的单核细胞而不是pDCs,需要TLR7而不是RLRs。在这种模型下,U1小型核糖核蛋白活化TLR7,尽管Fc受体对Ⅰ型干扰素的产生是必不可少的。
虽然,TLR7和TlR9在表达、位置、信号通路和靶基因表达上都是相似的,它们在小鼠自身免疫疾病发病机理中的作用是不同的。在MRLlpr/ lpr 小鼠上,TLR7缺陷导致更少对RNA相关抗原的自身抗体,并导致系统性免疫疾病如系统性红斑狼疮。但是,TLR9缺陷导致相关及效用。表明,TLR9介导的通路现在TLR7反应。
似乎自身免疫病也可由不合适的对自身核酸分子清除引起。人类和小鼠的狼疮样综合症都与编码血浆DNA酶Ⅰ的基因的边缘有关。缺乏溶酶体DNA酶Ⅱ的小鼠会在巨噬细胞中积累不完全消化的DNA,并有更高的Ⅰ型干扰素和TNF浓度,并形成自身免疫症如慢性关节炎。值得一提的是,DNA酶Ⅱ缺失的情况下Ⅰ型干扰素会增加而不依赖TLRs。编码修复核酸外切酶的基因的变异已在Aicardi-Goutières syndrome中发现,具有产生更多Ⅰ型干扰素有关的致命脑炎的特点。TREX1在细胞中的丢失导致来自复古元素的DNA的积聚,它负责诱导干扰素反应。编码核酸外切酶FEN1的基因突变的小鼠在凋亡细胞中也有更多的未消化DNA,并易患自身免疫病、慢性炎症和癌症。而且,编码RNA酶H2的成分SAMHD1的基因突变也与Aicardi-Goutières syndrome有关,还与抗病毒先天免疫反应的负调控有关。尽管这些不合适的核酸分子引起自身免疫的机制还所知甚少,但针对核酸的胞浆PRRs很可能参与了这一过程(图4)。
前景展望
在过去的十年中,在认识TLRs识别抗原和在宿主防御中的作用上,我们已经取得了巨大的进步。许多TLRs的结果分析阐明了TLR同二聚体或TLR异二聚体识别PAMP的机制,而且参与NF-κB, AP-1和IRF蛋白活化的许多信号分子也都已经被鉴定出来,并做了具体描述。目前研究的焦点是TLR信号的综合调控,包括转录后调控如泛素化、磷酸化和mRNA 的稳定性以及TLRs和它们的信号复合物的时空调控。而且,发现负调控的丢失与炎性疾病有直接关系,这表明药物抑制TLR反应可以有效的治疗这类疾病。
在一些非感染的疾病状态下的组织损伤可以活化TLR信号的发现,表明内源分子可充当TLR激动剂,尽管尚不清楚,这一反应对于维持稳态如组织修复是否有生物学意义,又或者这种识别仅仅是偶然现象。值得关注的是,微生物感染触发被修饰的内源分子的产生(如HMGB1、氧化磷脂、β防御素2和核酸),这些内源分子被TLRs或其它胞浆PRRs识别。这或许表示,这些内源分子和PAMPs一同充当通过TLRs和或PRRs激活先天免疫反应的佐剂,并在帮助抵抗微生物感染的获得性免疫中发挥关键作用。而且,已经越来越清楚的知道,在非免疫细胞中的胞浆PRR信号在促进DC成熟及随后特异抗原T细胞的活化中发挥作用。因此,在设计有效的疫苗佐剂时,应用可以刺激免疫和非免疫细胞的PAMPs和或内源激动剂或许会是有用的手段。
了解TLR活化中转录网络的复杂性并界定随后的免疫反应是当前研究的热点。系统生物学的手段是非常有用的,这一途径已成功预测了多种转录因子,如ATF3和C/EBPδ,是
IL-6转录的调节因子,这已经在敲除相关基因的细胞中得到证实。应用小干扰RNA来认识复杂的TLR转录网络的综合分析还只是刚刚开始。由于不同的细胞型、不同的TLR有不同的TLR信号,而且微生物抗原包含多个PAMPs,需要重视积累用各种TLR激动剂或抗原刺激的细胞型基因表达图谱的认识。为了全面了解不同PRRs在宿主防御抵抗感染中的协同作用,需要体内、体外和计算机模拟等各种手段的综合应用。
感想:本文中,作者介绍了TLRs的结构、类型、分布、功能及其配体的最新研究进展。重点阐述了各种TLRs介导的信号通路及其调节机制,介绍了其信号传导过程中的调节因子,及其影响因素。同时还介绍了非TLR胞浆模式识别受体及信号传导机制。最后,总结了TLRs 的研究现状,并对将来研究的方向做出了推断。
在我将来要从事的家禽遗传育种工作中,抗病育种长期受到研究者的青睐。但家禽由于其特殊的生理构造及连续的选育,而对病原菌的耐受性很差。TLRs是研究最为深入的一类模式识别受体,TLRs及其信号途径是连接先天性免疫和获得性免疫的桥梁,TLRs不仅参与了抗病毒、细菌及寄生虫感染早期先天性免疫应答,并在获得性免疫应答中也发挥作用。但相对于哺乳动物TLRs 的研究,有关鸡TLRs研究尚处于起步阶段。对鸡TLRs信号通路的深入了解将有助于我们进一步弄清禽类疾病相关微生物感染的发病机制,为抗病选育、相应疾病的治疗和疫苗研制等方面的研究提供新的切入点。
肿瘤常见信号通路
1 JAK-STAT 信号通路 1) JAK 与STAT 蛋白 JAK-STAT 信号通路是近年来发现的一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程。与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体、酪氨酸激酶JAK和转录因子STAT。 (1) 酪氨酸激酶相关受体( tyrosine kinase associated receptor ) 许多细胞因子和生长因子通过JAK-STAT 信号通路来传导信号,这包括白介素2?7 (IL-2?7 )、GM-CSF (粒细胞/巨噬细胞集落刺激因子)、GH (生长激素)、EGF (表皮生长因子)、PDGF (血小板衍生因子)以及IFN (干扰素)等等。这些细胞 因子和生长因子在细胞膜上有相应的受体。这些受体的共同特点是受体本身不具有激酶活性,但胞内段具有酪氨酸激酶JAK 的结合位点。受体与配体结合后,通过与之相结合的JAK 的活化,来磷酸化各种靶蛋白的酪氨酸残基以实现信号从胞外到胞内的转递。 (2) 酪氨酸激酶JAK ( Janus kinase ) 很多酪氨酸激酶都是细胞膜受体,它们统称为酪氨酸激酶受体( receptor tyrosine kinase, RTK ),而JAK 却是一类非跨膜型的酪氨酸激酶。JAK 是英文Janus kinase 的缩写,Janus 在罗马神话中是掌管开始和终结的两面神。之所以称为两面神激酶,是因为JAK既能磷酸化与其相结合的细胞因子受体,又能磷酸化多个含特定 SH2结构域的信号分子。JAK蛋白家族共包括4个成员:JAK1、JAK2、JAK3以及Tyk2,它们在结构上有7个JAK同源结构域(JAK homology domain, JH ),其中JH1结构域为激酶区、JH2结构域是“假”激酶区、JH6和JH7是受体结合区域。 (3) 转录因子STAT ( signal transducer and activator of transcription ) STAT 被称为“信号转导子和转录激活子”。顾名思义,STAT在信号转导和转录激活上发挥了关键性 的作用。目前已发现STAT家族的六个成员,即STAT1-STAT6。STAT蛋白在结构上可分为以下几个功能区段:N-端保守序列、DNA结合区、SH3结构域、SH2结构域及C-端的转录激活区。其中,序列上最保守和功能上最重要的区段是SH2结构域,它具 有与酪氨酸激酶Src的SH2结构域完全相同的核心序列“ GTFLLRFSS ”。 2) JAK-STAT 信号通路 与其它信号通路相比,JAK-STAT 信号通路的传递过程相对简单。信号传递过程如下:细胞因子与相应的受体结合后引起受体分子的二聚化,这使得与受体偶联的JAK激酶相互接近并通过交互的酪氨酸磷酸化作用而活化。JAK激活后催化受体上的酪氨酸残 基发生磷酸化修饰,继而这些磷酸化的酪氨酸位点与周围的氨基酸序列形成“停泊位
Toll样受体信号通路的研究进展
Toll样受体信号通路的研究进展 摘要Toll样受体(Toll-like receptor,TLR)是近年来发现的一类模式识别受体,通过识别病原相关分子模式(pathogen-associated molecular pattern,PAMP)激活天然免疫。而髓样分化因子(myeloid differentiation factor 88,MyD88)是TLR信号通路中的一个关键接头分子,在传递上游信息和疾病发生发展中具有重要的作用。本文对Toll样受体、髓样分化因子88的分子结构和基本功能,及Toll样受体的信号传导通路进行了综述。 关键词Toll样受体;髓样分化因子88;信号通路;负调控机制 免疫系统识别“非我”和“自我”的过程是依赖于不同的受体来完成的,作为先天性免疫系统的重要组成部分及连接获得性免疫与先天性免疫的“桥梁”, TLRs 是生物的一种模式识别受体(pattern recognition receptor, PRR),它主要通过识别病原相关分子模式PAMPs来启动免疫反应。而MyD88是Toll受体信号通路中的一个关键接头分子,是第一个被鉴定的含TIR结构域的接头蛋白分子,在传递上游信息和疾病发生发展中具有重要的作用。 1TLR的结构与基本功能 Toll样受体一词来自对果蝇的研究,是决定果蝇背腹分化的基因所编码的一种跨膜受体蛋白,同时还参与果蝇的免疫反应,具有介导抗真菌感染信号转导的功能[1]。后来在哺乳动物也发现有与Toll受体同源的受体分子,统称为称为Toll 样受体TLRs。 TLRs是广泛分布在免疫细胞尤其非特异免疫细胞以及某些体细胞表面的一类模式识别受体,它们可以直接识别结合某些病原体或其产物所共有的高度保守的特定分子结构,即病原相关分子模式。迄今为止,已经发现哺乳动物至少有13种toll样受体,其中人的toll样受体鉴定出11种(TLR1-TLR11) [2]。TLRs识别的配基各不相同,其中TLR1-TLR5的结构已被确定,但只有TLR2与TLR4的功能被部分揭示。TLR4主要介导G-菌感染后LPS的信号转导,而TLR2主要介导G+感染后脂蛋白、脂多肽等的信号转导。它们都最终导致该转录因子的转位与相应免疫基因的活化而转录,释放前炎症因子及辅助刺激分子起到调节炎症反应的作用,从而提示TLRs可能在先天性免疫系统中起重要作用[3-4]。 TLRs家族成员具有相似的结构特征。它们均为Ⅰ型跨膜受体,由胞外区、跨膜区和胞内区3个功能区组成。胞外区序列差异大,是与配体结合的特异部位,主要包括十几至二十几个串联的富亮氨酸重复基序(leucine-rich repeats, LRRs),LRR
脂多糖模式识别受体的研究进展
[收稿日期]2000-10-30; [修回日期]2000-11-14 [基金项目]国家重点基础研究发展规划资助项目(G1999054203) [作者简介]顾长国(1970-),男,福建建宁县人,助理研究员,硕士,主要从事创伤感染免疫研究。 T el:(023)68757431; E -mail :g ucg @china .com [文章编号]1000-8861(2001)02-0150-03 脂多糖模式识别受体的研究进展 顾长国综述,李 磊审校 (第三军医大学大坪医院外研所一室,重庆400042) [摘 要] G -细菌的脂多糖(L PS)是重要的病原体相关模式分子。P AM Ps 均可被动物作为外来分子进行识别。LP S 能 激发机体细胞因子IL -1、T N F- 等活性分子的合成,对感染具有十分重要的作用。L PS 是通过什么受体怎样将信号传入免疫细胞并启动免疫反应的,人们一直都不十分清楚。近年来,一种名为T o ll 蛋白的发现,使人们对机体识别LP S 机制的认识向前跨进了一大步。本文试对该模式识别受体的研究进展做一综述。 [关键词] 脂多糖;模式识别受体;T o ll-like receptor s(T L Rs)[中图分类号] R 392.1 [文献标识码] A Recent advance in research of pattern recognition receptors for LPS GU Chang-guo (First Dep artment of Institutes of Sur gical Research ,Dap ing H osp ital ,T hird M ilitary M edical University ,Chongqing 400042,China ) [Abstract ] L ipopolysaccharide (L PS )of G ram -neg ative bacteria is an import ant pat ho gen-associated molecular pat ter ns (P A M Ps).T hey can be detected as ex og eno us molecules.L P S plays an impo rtant role in infection and trig ger innate immune r e-sponse.But it is no t well known how it w or ks.U nv eiling of T oll-like receptors,a t ype Ⅰtransmembrane pr otein,has m ade a gr eat pr ogr ess in understanding t he mechanism of recognitio n of L PS.Here,we try to introduce recent resear ch advances o f this po tential patter n r ecog nition r ecepto rs(PR R). [Key words ] lipopo lysaccharide(L P S);pattern recognit ion receptor s(P RR );T oll-like r eceptor s(T L Rs) 微生物细胞壁的组成成分是先天性免疫反应的高效活化分子。这些分子如G -细菌的脂多糖(lipopoly sacchar ide,L PS )、G +细菌的肽聚糖(pept idog ly can )、脂磷壁酸(lipot ei-choic acid ,T LA )以及真菌的甘露糖(mannans )等称为病原体相关的模式分子(pathog en-associated m olecular patterns,PA M P s)。与之相对应的模式识别受体(patter n r ecog nition receptor s ,P RR )这一概念最早是由Janew ay 在1992年提出的[1]。P AM Ps 均可被动物作为外来分子进行识别。PA M P s 能激发机体细胞因子如I L -1、T N F- 等以及其他活性分子的合成,对感染具有十分重要的作用。但是细胞因子的过度活化可以引起脓毒症休克,是细菌感染患者死亡的首要原因。因此模式识别受体在先天性免疫中居于重要地位,通过模式识别受体机体能区别病原体与自身组织,这是免疫反应的起点和根本特征。但过去的研究一直都没有发现能识别PA M P s 特别是L P S 并引起上述反应的模式识别受体。近年来发现的T oll-like receptors(T LR s)是在研究L PS 作用机制方面获得的重要进展,对深入了解模式识别受体作用机制, 研究先天性免疫反应的传入途径是一个重大突破。1 LPS 作用机制的研究 L PS 可以说是先天性免疫最强的刺激剂。 70年代人们普遍认为L PS 要发挥作用,必须先插入生物膜脂质双分子层或通过受体作用被吞噬,但这样的猜想一直无法得以证实。Cout inbo 发现C3H/HeJ 小鼠对L PS 无反应性,并将其原因归结为位于常染色体的1个等位基因位点lps 的突变。但是C 3H /HeJ 小鼠对G +细菌的反应却正常,由L PS 介导产生的细胞因子等方面也是正常的。这种表型上的差异可以说是因为对L PS 识别的缺陷造成的。lpsd 动物实验的研究可以得到一个结论:在哺乳动物存在对微生物的天然识别机制,这种机制识别的范围可以粗略的定义为G -细菌。这个天然识别机制的在对感染的耐受方面起重要作用。对lps d 动物的研究使人们认识到对L PS 的识别机制是先天性免疫的重要环节,也必然存在一条信号传导途径能将L PS 的作用引入胞内,而且lps d 纯合子在L PS 作用时信号将完全阻断。可以进一步推 论lps 编码的产物能识别L PS 并引起对G -菌感染的快速反 ?150? 免疫学杂志 第17卷 第2期2001年3月 IM M U NO LO GICA L JO U RN A L Vo l 17N o 2M ar 2001
常见的信号通路
1 JAK-STAT信号通路 1) JAK与STAT蛋白 JAK-STAT信号通路是近年来发现的一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程。与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体、酪氨酸激酶JAK和转录因子STAT。 (1) 酪氨酸激酶相关受体(tyrosine kinase associated receptor) 许多细胞因子和生长因子通过JAK-STAT信号通路来传导信号,这包括白介素2?7(IL-2?7)、GM-CSF(粒细胞/巨噬细胞集落刺激因子)、GH(生长激素)、EGF(表皮生长因子)、PDGF (血小板衍生因子)以及IFN(干扰素)等等。这些细胞因子和生长因子在细胞膜上有相应的受体。这些受体的共同特点是受体本身不具有激酶活性,但胞内段具有酪氨酸激酶JAK的结合位点。受体与配体结合后,通过与之相结合的JAK的活化,来磷酸化各种靶蛋白的酪氨酸残基以实现信号从胞外到胞内的转递。 (2) 酪氨酸激酶JAK(Janus kinase) 很多酪氨酸激酶都是细胞膜受体,它们统称为酪氨酸激酶受体(receptor tyrosine kinase, RTK),而JAK却是一类非跨膜型的酪氨酸激酶。JAK是英文Janus kinase的缩写,Janus在罗马神话中是掌管开始和终结的两面神。之所以称为两面神激酶,是因为JAK既能磷酸化与其相结合的细胞因子受体,又能磷酸化多个含特定SH2结构域的信号分子。JAK蛋白家族共包括4个成员:JAK1、JAK2、JAK3以及Tyk2,它们在结构上有7个JAK同源结构域(JAK homology domain, JH),其中JH1结构域为激酶区、JH2结构域是“假”激酶区、JH6和JH7是受体结合区域。 (3) 转录因子STAT(signal transducer and activator of transcription)STAT被称为“信号转导子和转录激活子”。顾名思义,STAT在信号转导和转录激活上发挥了关键性的作用。目前已发现STAT家族的六个成员,即STAT1-STAT6。STAT蛋白在结构上可分为以下几个功能区段:N-端保守序列、DNA结合区、SH3
TOLL样受体
TLR结构:TOLL样受体(TLR)为I型跨膜蛋白,其胞外段为富含亮氨酸重复序列,参与配体识别;胞内段含有保守的TIR (TOLL样/IL一IR)结构域,招募衔接分子如MYD88、TIRAP、TRIF、TRAM{1}进行信号转导。 TLR识别配体:TLR是结合病原微生物成分的受体,其配体包括合成的激动药、微生物产物、内源性配体{1}其所识别的病原微生物成分包括脂多糖(lipoPolysaeeharide,LpS)、革兰氏阳性细菌的肤聚糖(peptidoglyean,pGN)、脂磷壁酸(liPoteiehoieaeid,LTA)、脂阿拉伯甘露聚糖(11-poarabinomannan,LAM)等。 TLR分类:在人类已发现10种TLR(TLRI一TLmo),表达于参与天然免疫的细胞上,不同的TLR在不同细胞表面有不同的表达,其所识别的配体亦不同。髓系DC表达TOLL 样受体1-6、8,而浆系DC表达TOLL样受体7、9。与DC成熟关系密切的是TLR2、TLR4。其中TLR2识别脂蛋白类,肽多糖类如革兰氏阳性细菌的肤聚糖(peptidoglyean,pGN)、脂磷壁酸(liPoteiehoieaeid,LTA)。而TLR4识别LPS、OK432等。 TLR与DC成熟的关系:{2}{5}
TLR信号转导机制:{3}
TLR受体激动药在肿瘤微环境下的免疫调节作用:{1} TLR基因定位:{4} 特异性引物序列: TLR2(forward GCAAACGCTGTTCTGCTCAG) (reverse AG GCGTCTCCCTCTA TTGTA TT) TLR4 (forward ATGGCATGGCTTACACCACC) (reverse GA GGCCAA TTTTGTCTCCACA)
模式识别受体与肿瘤微环境研究进展_顾炎
中国肿瘤生物治疗杂志http ://www.biother.org Chin J Cancer Biother ,Apr.2015,Vol.22,No.2 doi :10.3872/j.issn.1007-385X.2015.02.002 ·院士论坛· 模式识别受体与肿瘤微环境研究进展 顾炎,曹雪涛(第二军医大学免疫学研究所暨医学免疫学国家重点实验室,上海200433) [基金项目]国家重点基础研究发展计划(973计划)资助项目(No.2011CB965202);国家自然科学基金青年科学基金资助项目(No.31400757)。Project supported by the National Key Basic Research Program of China (No.2011CB965202),and the National Natural Science Founda-tion for the Youth (No.31400757 ) 顾炎博士,讲师,任职于第二军医大学免疫学研究所暨医学免疫学国家重点实验室。2008年毕业于 第二军医大学临床医学系,同年进入医学免疫学国家重点实验室师从曹雪涛院士,2013年获得医学免疫学博士学位,并留校任教。主要从事肿瘤免疫逃逸的细胞与分子调控研究,研究重点为免疫细胞参与肿瘤负向免疫调控机制。博士课题揭示了肿瘤驯化的B 淋巴细胞对乳腺癌转移的促进功能,揭示了B 细胞及其产生的抗体参与肿瘤免疫逃逸的新的作用机制,并发现预测乳腺癌淋巴结转移以及患者预后判断的重要血清学指标。目前在研课题主要关注天然免疫细胞及Toll 样受体在肿瘤肺转移中新的作用及其机制。获国家自然科学基金青年基金一项,参与多项“973”、“863”、“科技重大专项”等国家级课题项目,研究成果发表在Hepatology 、 J Immunol 、Oncoimmunology 等杂志。E-mail :guyan_84@163.com 曹雪涛教授,博士生导师, 中国工程院院士。现任中国医学科学院院长、第二军医大学免疫学研究所所长、医学免疫学国家重点实验室主任,全球慢性疾病防控联盟主席、亚大地区免疫学联盟主席,中国免疫学会秘书长、国家“863计划”现代医学主题专家组组长、国家“973计划”免疫学项目首席科学家、国务院学位评议委员会学科评议基础医学组召集人。任《中国肿瘤生物治疗杂志》主编,Cell Mol Immunol 杂志共同主编,J Mol Med 、Gene Ther 、Cancer Immunol Res 等杂志的副主编,Cell 、Ann Rev Immu-nol 、Sci Transl Med 、eLife 等杂志的编委。主要从事天然免疫识别与免疫调节的基础研究和疾病免疫治疗的应用研究,以通信作者身份在Science 、Cell 、Nat Immunol 等杂志发表SCI 论文226篇,论文被SCI 他引6000余次;主编和共同主编学术专著8部,参编11部;获国家发明专利16项。培养的12名博士的学 位论文获评“全国百篇优秀博士学位论文”。E-mail :caoxt@immunol.org [摘 要]肿瘤微环境的组成与肿瘤发生发展的关系备受瞩目,免疫系统参与肿瘤微环境形成并在其中发挥重要作用,天然 免疫细胞对肿瘤的免疫监视以及免疫耐受的形成具有双向功能。模式识别受体(pattern recognition receptors , PRRs )是天然免疫细胞识别病毒、细菌等病原体以启动免疫与炎症过程的受体,在肿瘤免疫中也发挥双向的调控功能,其既能维持宿主寄生菌群平衡、清除死亡或突变细胞以抑制肿瘤发生,又能诱导慢性炎症、形成炎性微环境以促进肿瘤发生;既能识别危险信号启动天然免疫杀伤及后续的获得性免疫应答以抑制肿瘤进程,又能识别肿瘤释放的内源性配体以促进抑制性细胞亚群和细胞因子的产生,进而诱导肿瘤的免疫耐受与免疫抑制。此外,多种肿瘤细胞亦表达多种PRRs ,肿瘤细胞本身PRR通路参与了肿瘤的发生、发展。本文将阐述肿瘤微环境中PRRs 及其配体表达的特点,重点分析PRRs 在肿瘤免疫调控中发挥的双向调控功能,以期为肿瘤免疫微环境形成的认识及肿瘤免疫治疗的设计提供新的视角。[关键词]肿瘤;肿瘤微环境;模式识别受体;天然免疫;免疫调控;免疫治疗[中图分类号]R730.23;R730.54;R392.11 [文献标志码]A [文章编号]1007- 385X (2015)02-0143-08Recent progress in the research on pattern recognition receptors and tumor microenvironment Gu Yan ,Cao Xuetao (National Key Laboratory of Medical Immunology &Institute of Immunology ,Second Military Medi-cal University ,Shanghai 200433,China ) [Abstract ]Tumor microenvironment has attracted significant research attentions worldwide.It is now generally accepted · 341·
cAMP信号通路
cAMP信号通路 信号分子:1.激素 2.局部介质3.神经递质 受体:G蛋白偶联受体 胞内应答过程:激素→G蛋白耦联受体→G蛋白→腺苷酸环化酶→cAMP→依赖cAMP的蛋白激酶A→基因调控蛋白→基因转录 举例:1.多发性骨髓瘤:通过调变细胞内环腺苷酸浓度可以诱导多种肿瘤细胞增殖阻滞和凋亡,成为肿瘤治疗新途径。 2.肝损伤:对乙酰氨基酚致药物性肝脏损伤可能与cAMP-PKA 信号通路有关。 3.研究人员已经确定了这其中的机制,现在,一种能抑制Epac的新的候选药物——称为ESI Epac特异性抑制剂,也已经被证明能够保护正常小鼠免受致命性立克次氏体感染。目前,研究人员正在设计第二代ESI——更有效,即使在最高剂量也无毒。也有来自预备试验的迹象表明,ESI能够保护动物抗击一些致命的病毒感染。 磷脂酰肌醇信号通路 信号分子:1.激素 2.局部介质3.神经递质 受体:酶耦联型受体 胞内应答过程:Ca2+活化各种Ca2+结合蛋白引起细胞反应,钙调素(calmodulin,CaM)由单一肽链构成,具有四个钙离子结合部位。结合钙离子发生构象改变,可激活钙调素依赖性激酶(CaM-Kinase)。细胞对Ca2+的反应取决于细胞内钙结合蛋白和钙调素依赖性激酶的种类。 IP3信号的终止是通过去磷酸化形成IP2,或被磷酸化形成IP4。Ca2+由质膜上的Ca2+泵和Na+-Ca2+交换器将抽出细胞,或由内质网膜上的钙泵抽进内质网 DG通过两种途径终止其信使作用:一是被DG-激酶磷酸化成为磷脂酸,进入磷脂酰肌醇循环;二是被DG酯酶水解成单酯酰甘油。由于DG代谢周期很短,不可能长期维持PKC活性,而细胞增殖或分化行为的变化又要求PKC长期活性所产生的效应。现发现另一种DG生成途径,即由磷脂酶催化质膜上的磷脂酰胆碱断裂产生的DG,用来维持PKC的长期效应。 举例:1.肿瘤治疗:该通路调节肿瘤细胞的增殖和存活,其活性异常不仅能导致细胞恶性转化,而且与肿瘤细胞的迁移、黏附、肿瘤血管生成以及细胞外基质的降解等相关。 2.肝癌:PIK3R1在肝癌组织中表达上调,PIK3R1可能通过激活PI3K/AKT信号通路促进HepG2细胞的增殖. 生物技术15-1 曹文祥
integrin review 整合素受体信号通路综述
Rheumatol Int DOI 10.1007/s00296-014-3137-5 Role of integrins and their ligands in osteoarthritic cartilage Jian Tian · Fang?Jie Zhang · Guang?Hua Lei Received: 25 May 2014 / Accepted: 17 September 2014 ? Springer-Verlag Berlin Heidelberg 2014 [1]. Radiographic evidence of OA occurs in the majority of people by 65 years of age, and among them about 80 % in people who aged over 75 years [2]. However, the pathogen-esis of this disease is not fully elucidated. Cartilage damage is one of the major pathological changes in OA. Articular cartilage is an avascular, a neu-ral, alymphatic, and viscoelastic connective tissue that functions autonomously to bear loads and provide almost friction-free movement of diarthrodial joints [3]. Chondro-cytes, the only cell population of adult articular cartilage, are strongly involved in maintaining the dynamic equi-librium between synthesis and degradation of the extra-cellular matrix (ECM) [4]. Collagens represent the major structural components of the articular cartilage. Cartilage is made up of two main ECM macromolecules: type II collagen and aggrecan, a large aggregating proteoglycan [5, 6]. Cartilage destruction is thought to be mediated by two main enzyme families: the matrix metalloproteinases (MMPs) are responsible for the cartilage collagen break-down, whereas enzymes from disintegrin and metallopro-teinase domain with thrombospondin motifs (ADAMTS) family mediate cartilage aggrecan loss [7]. Activation of biochemical pathways involves the production of proin-flammatory cytokines, inflammation, degradation of the ECM by MMPs and ADAMTS, and cessation of ECM syn-thesis via dedifferentiation and apoptosis of chondrocytes [8, 9]. Therefore, the ECM is a vital cellular environment, and interactions between the cell and ECM are important in regulating many biological processes, which include cell growth, differentiation, and survival [10, 11]. Cell–matrix interactions control cell function and behav-ior by signal transduction through a variety of cell sur-face receptors. The integrins are the major family of ECM receptors, which can transmit information from the matrix to the cell. Integrin binding of ECM ligands results in the Abstract Osteoarthritis (OA) is a degenerative disease, which is characterized by articular cartilage destruction, and mainly affects the older people. The extracellular matrix (ECM) provides a vital cellular environment, and interactions between the cell and ECM are important in reg-ulating many biological processes, including cell growth, differentiation, and survival. However, the pathogenesis of this disease is not fully elucidated, and it cannot be cured totally. Integrins are one of the major receptors in chondro-cytes. A number of studies confirmed that the chondrocytes express several integrins including α5β1, αV β3, αV β5, α6β1, α1β1, α2β1, α10β1, and α3β1, and some integrins ligands might act as the OA progression biomarkers. This review focuses on the functional role of integrins and their extracellular ligands in OA progression, especially OA car-tilage. Clear understanding of the role of integrins and their ligands in OA cartilage may have impact on future develop-ment of successful therapeutic approaches to OA.Keywords Chondrocyte · Integrin · Fibronectin · Tenascin C · Osteopontin · Osteoarthritis · Cartilage Introduction Osteoarthritis (OA) is a degenerative disease and is char-acterized by articular cartilage destruction along with changes occurring in other joint components including bone, menisci, synovium, ligaments, capsule, and muscles Rheumatology INTERNATIONAL J. Tian · F.-J. Zhang · G.-H. Lei (*) Department of Orthopaedics, Xiangya Hospital, Central South University, No. 87 Xiangya Road, Changsha 410008, Hunan, China e-mail: gh.lei9640@https://www.360docs.net/doc/f817471227.html,; lgh9640@https://www.360docs.net/doc/f817471227.html,
Toll样受体信号通路图
Toll样受体信号通路图 TLR家族成员(TLR3除外)诱导的炎症反应都经过一条经典的信号通路(图1),该通路起始于TLRs的一段胞内保守序列—Toll/IL-1受体同源区(Toll/IL-1receptorhomologousregion,TIR).TIR可激活胞内的信号介质—白介素1受体相关蛋白激酶(IL-1Rassociatedkinase,IRAK)IRAK-1和IRAK-4、肿瘤坏死因子受体相关因子6(TNFR-associatedfactor6,TRAF-6)、促分裂原活化蛋白激酶(mitogenactivatedproteinkinase,MAPK)和IκB激酶(IκBkinase,IκK),进而激活核因子κB(nuclearfactorκB,NF-κB),诱导炎症因子的表达。 Toll-liker Receptor Signaling 本信号转导涉及的信号分子主要包括: CD14,MD-2,TRAM,TRIF,TIRAP,MyD88,TLR1,TLR2,TLR3,TLR4,TLR5,TLR6,TLR7,TLR8,TLR9,IRAK-1,IRAK-2,IRAK-4,IRAK-M,TRAF6,TRIAD3A,ST2L,SOCS1,RIG-I,FADD,TOLLIP,RIP1,A20,UEV1A,Ubc13,ECSIT,MEKK-1,TAK1,
TBK1,MKK3/6,p38,TAB1/2,MKK4/7,JNK,IKKα,IKKβ,IKKγ,IKKε,NEMO,IκBα,NF-κB,p65/RelA,Casp-8,IRF-3,IRF-7,MA VS等
细胞受体及重要的细胞信号转导途径
细胞受体类型、特点 及重要的细胞信号转导途径 学院:动物科学技术学院 专业:动物遗传育种与繁殖 姓名:李波
学号:2015050509
目录 1、细胞受体类型及特点 (4) 1.1离子通道型受体 (4) 1.2 G蛋白耦联型受体 (4) 1.3 酶耦联型受体 (5) 2、重要的细胞信号转导途径 (5) 2.1细胞内受体介导的信号传递 (5) 2.2 G蛋白偶联受体介导的信号转导 (6) 2.2.1激活离子通道的G蛋白偶联受体所介导的信号通路 (7) 2.2.2激活或抑制腺苷酸环化酶的G蛋白偶联受体 (7) 2.2.3 激活磷脂酶C、以lP3和DAG作为双信使 G蛋白偶联受体介导的信号通 路 (8) 2.2 酶联受体介导的信号转导 (9) 2.2.1 受体酪氨酸激酶及RTK-Ras蛋白信号通路 (10) 2.2.2 P13K-PKB(Akt)信号通路 (10) 2.2.3 TGF-p—Smad信号通 (11) 2.2.4 JAK—STAT信号通路 (12)
1、细胞受体类型及特点 受体(receptor)是一种能够识别和选择性结合某种配体(信号分子)的大分子物质,多为糖蛋白,一般至少包括两个功能区域,与配体结合的区域和产生效应的区域,当受体与配体结合后,构象改变而产生活性,启动一系列过程,最终表现为生物学效应。受体与配体问的作用具有3个主要特征:①特异性;②饱和性;③高度的亲和力。 根据靶细胞上受体存在的部位,可将受体分为细胞内受体(intracellular receptor)和细胞表面受体(cell surface receptor)。细胞内受体介导亲脂性信号分子的信息传递,如胞内的甾体类激素受体。细胞表面受体介导亲水性信号分子的信息传递,膜表面受体主要有三类:①离子通道型受体(ion—channel—linked receptor);②G蛋白耦联型受体(G—protein —linked receptor);③酶耦联的受体(enzyme—linked recep—tor)。第一类存在于可兴奋细胞。后两类存在于大多数细胞,在信号转导的早期表现为激酶级联事件,即为一系列蛋白质的逐级磷酸化,借此使信号逐级传送和放大。 1.1离子通道型受体 离子通道型受体是一类自身为离子通道的受体,即配体门通道(1igand—gated channel),主要存在于神经、肌肉等可兴奋细胞,其信号分子为神经递质。神经递质通过与受体的结合而改变通道蛋白的构象,导致离子通道的开启或关闭,改变质膜的离子通透性,在瞬间将胞外化学信号转换为电信号,继而改变突触后细胞的兴奋性。如:乙酰胆碱受体以三种构象存在,两分子乙酰胆碱的结合可以使之处于通道开放构象,但该受体处于通道开放构象状态的时限仍十分短暂,在几十毫微秒内又回到关闭状态。然后乙酰胆碱与之解离,受体则恢复到初始状态,做好重新接受配体的准备。离子通道型受体分为阳离子通道,如乙酰胆碱、谷氨酸和五羟色胺的受体,和阴离子通道。 1.2 G蛋白耦联型受体 三聚体GTP结合调节蛋白(trimeric GTP—binding regulatory protein)简称G蛋白,位于质膜胞质侧,由a、p、-/三个亚基组成,a和7亚基通过共价结合的脂肪酸链尾结合在膜上,G蛋白在信号转导过程中起着分子开关的作用,当a亚基与GDP结合时处于关闭状态,与GTP结合时处于开启状态,“亚基具有GTP酶活性,能催化所结合的ATP 水解,恢复无活性的三聚体状态,其GTP酶的活性能被RGS(regulator of G protein signaling)增强。RGS也属于GAP(GTPase activating protein)。 G蛋白耦联型受体为7次跨膜蛋白(图10—6),受体胞外结构域识别胞外信号分子并与之结合,胞内结构域与G蛋白耦联。通过与G蛋白耦联,调节相关酶活性,在细胞内
TOLL样受体7(TLR7)增殖分化信号通路论文
TOLL样受体7(TLR7)增殖分化信号通路论文 【提示】本文仅提供摘要、关键词、篇名、目录等题录内容。为中国学术资源库知识代理,不涉版权。作者如有疑义,请联系版权单位或学校。 【摘要】目的探讨TLR7的激活对HaCaT细胞增殖与分化的影响及其可能的机制。方法培养HaCaT细胞,以不同剂量的TLR7配体Gardiquimod经不同的时间体外刺激HaCaT细胞,MTT及流式细胞术分析TLR7的激活对HaCaT细胞增殖的影响。以不同剂量的TLR7配体Gardiquimod经不同的时间体外刺激HaCaT细胞,加入氯化钙诱导HaCaT细胞分化,Western-Blot分析HaCaT细胞的分化Markers(颗粒层:Keratin1,基底层:Keratin5和棘层:Involucrin)并以此分析TLR7的激活对氯化钙诱导HaCaT细胞分化的影响。 Western-blotting分析TLR7在HaCaT细胞中激活的信号通路 PI3K-AKT和RAS-MAPK等。在TLR7配体Gardiquimod处理HaCaT细胞前1h,分别加入特异性阻断剂(PD98059及LY2940002)阻断TLR7配体Gardiquimod激活的相关信号通路,然后分析阻断剂对TLR7配体Gardiquimod调控HaCaT细胞增殖及分化影响,从而探讨PI3K-AKT 和RAS-MAPK信号通路在TLR7配体Gardiquimod对HaCaT细胞增殖及分化调控中的作用。结果MTT及流式细胞分析结果显示:TLR7配体Gardiquimod促进HaCaT细胞增殖,且具有时间及剂量依赖性;TLR7配体Gardiquimod能够抑制氯化钙诱导的HaCaT细胞分化markers (Keratin1及Involucrin)的表达,存在时间效应及剂量效应;信号通路分析揭示TLR7配体Gardiquimod能够增加ERK1/2和MAPK的水平;阻断剂的研究发现TLR7配体Gardiquimod部分依赖PI3K-AKT
toll样受体信号通路
Toll 样受体(TLRs)是一个模式识别受体家族,它们在进化上高度保守,从线虫到哺乳 动物都存在TLRs,目前在哺乳动物中已发现 12 个成员[1].TLRs 主要表达于抗原递 呈细胞及一些上皮细胞,为玉型跨膜蛋白,胞外区具有富含亮氨酸的重复序列,能够 特异识别病原微生物进化中保守的抗原分子———病原相关分子模式 (pathogen-associatedmolecular patterns, PAMPs)[2].为了有效地抵抗入侵的病原体,机体需要对多种 PAMPs 产生适当的免疫应答,TLRs 可以通过识别 PAMPs 诱发抵抗病原体的免疫反应.而且 TLRs 也参与识别有害的内源性物质.TLRs 的激活可诱导很强的免疫反应,有利于机体抵抗病原体感染或组织损伤,但是过度的免疫反应也会带来不利影响,如产生内毒素休克、自身免疫性疾病等.为了保证 TLRs 介导正确的免疫应答,机体 存在精密的负调控机制,及时抑制 TLRs 信号,维持机体的免疫平衡[3]TLR 家族成员(TLR3 除外)诱导的炎症反应都经过一条经典的信号通路(图 1),该通路起始于TLRs 的一段胞内保守序列———Toll/IL-1 受体同源区(Toll/IL-1 receptor homologous region,TIR).TIR可激活胞内的信号介质———白介素 1 受体相关蛋白激酶 (IL-1R associated kinase, IRAK) IRAK-1 和IRAK-4、肿瘤坏死因子受体相关因子 6(TNFR-associated factor 6, TRAF-6)、促分裂原活化蛋白激酶(mitogen activated protein kinase,MAPK)和 I资B激酶 (I资B kinase, I资K ),进而激活核因子资 B(nuclear factor 资B,NF-资B),诱导炎症因子的表达.TLRs 信号通路上的许多接头蛋白都具有 TIR结构域:髓系分化因子 88(myeloid differentiationfactor 88, MyD88)、MyD88- 接头蛋白相似物(MyD88-adaptor like,Mal)、含有 TIR 结构能诱导干扰 素茁的接头分子 (TIR domain-containingadaptor inducing interferon 茁,TRIF)、TRIF 相关接头分子(TRIF-related adaptor molecule,TRAM)和SARM (sterile 琢 and armadillo motif-containingprotein)[4].它们参与 TLRs 所介导的信号转导,其中 MyD88 最重要,参与了除 TLR3 外所有 TLRs介导的信号转导.MyD88 首先通过 TIR 与 TLRs 相结合,接着募集下游信号分子 IRAK-4,IRAK-4 磷酸化激活IRAK-1,随后 活化 TRAF6.活化的 TRAF6 具有泛素连接酶(E3)的活性,能够结合泛素结合酶(E2),进而泛素化降解 IKK-酌.这种泛素化降解可以活化TGF-茁激酶(TGF-茁 activated kinase 1, TAK1) 和TAK1 结合蛋白 (TAK1 binding protein, TAB1、TAB2、 TAB3).活化的 TAK1 会催化 IKK-茁磷酸化,最终激活 NF-资B,促使炎症因子的表达.除了共同的 NF-资B 激活通路,不同的 TLRs 还存在着其特有的信号通路,一些TLRs 具有募集 Mal、TRAM 和 TRIF 的作用.不同的接头分子在信号传导中发挥的作 用不同[5],TRIF 在脂多糖(LPS)激活的 TLR4 途径和 Poly(I∶C)激活的 TLR3 途径中都起到了重要的作用,而 TRAM 仅在 TLR4 的途径中发挥作用.TLRs 的激活是一把双刃剑,它可以通过刺激先天性免疫应答和提高获得性免疫反应来保护机体,但是它所引 起的持续性炎症反应也会对机体产生损伤,自身免疫、慢性炎症和感染性疾病都与它 有一定关系.例如LPS 持续刺激TLR4 就可以引起严重的败血病和感染性休克,此外,类风湿性关节炎、慢性阻塞性肺心病、结肠炎、哮喘、心肌病、狼疮和动脉粥样硬化