全基因组重亚硫酸盐测序【最新版】

全基因组重亚硫酸盐测序
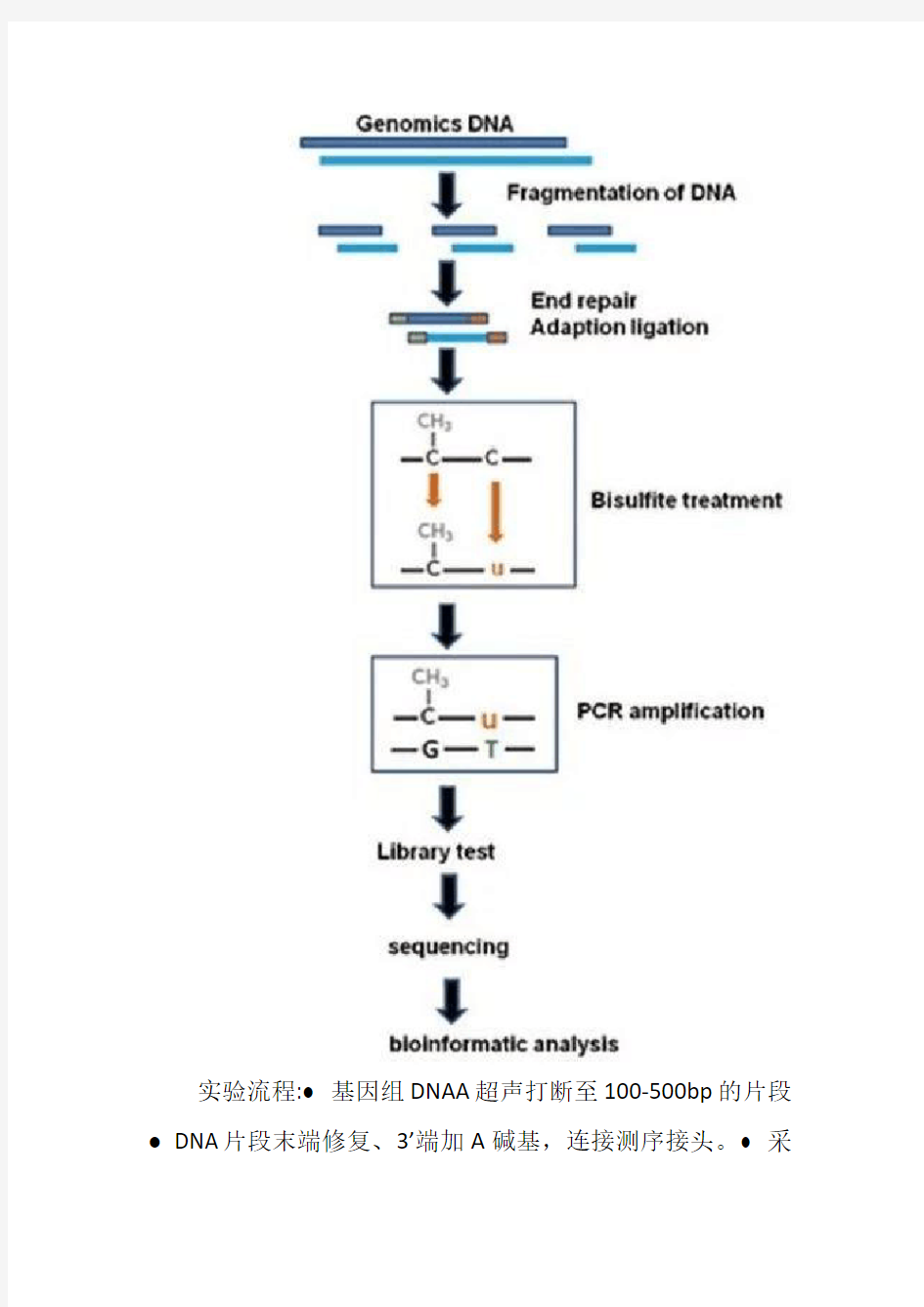
表观遗传学研究已经证实了特定基因区域的DNA甲基化修饰对于染色体构象、基因表达调控机制有着重要影响,而全基因组DNA甲基化研究将是表观基因组学最为关注的内容之一。Bisulfite处理能够将基因组中未发生甲基化的C 碱基转换成U,进行PCR扩增后变成T,与原本具有甲基化修饰的C碱基区分开来,再结合高通量测序技术,可绘制单碱基分辨率的全基因组DNA甲基化图谱。特定物种的高精确度甲基化修饰模式的分析,必将在表观基因组学研究中具有里程碑式的意义,并且为细胞分化、组织发育等基础机制研究,以及动植物育种、人类健康与疾病研究奠定基础。技术优势:■单碱基精确度:精确分析每一个C碱基的甲基化状态。■里程碑式的研究:特定物种的表观基因组学研究的重要内容,适用于所有具有精确基因组图谱的物种。
实验流程:● 基因组DNAA超声打断至100-500bp的片段● DNA片段末端修复、3’端加A碱基,连接测序接头。● 采
用EZ DNA Methylattion-Gold kit 进行Bisulfite 处理● 脱盐处理,PCR扩增后进行文库片段大小选择。● 合格的文库用于上机测序。
信息分析流程图:
生物信息分析:1. Data Clean测序结果进行去污染,去接头处理。根据测序产生的序列文件*.fq 统计read长度,read 数量,数据产量。
2. 标准信息分析2.1 Bisulfite-seeq 序列与参考序列的比对在信息分析过程中,首先将每一对reads中正链reads上的C碱基转换为T碱基,而反链reads中的G碱基转换为A 碱基。在此基础上使用SOAP软件,将reads与参考基因组序列进行比对,唯一比对reads将用于甲基化信息的分析。数据比对统计结果如下:
2.2 C碱基测序深度的累积分布甲基化C碱基在基因组上的分布包含三种形式(CG, CHG和CHH,其中H代表A 或T 或C碱基)。下述图表中反映了三种不同分布类型的C碱基的测序深度累积分布。其中横轴表示C碱基测序深度,纵轴表示一定测序深度下C碱基的累积比例( copynum <=1.5即uniquely>
碱基测序深度的累积分布图
2.3 不同reads测序深度下的基因组覆盖度横轴表示测序深度,纵轴表示特定测序深度下所对应的基因组覆盖度。
2.4 计算C碱基的甲基化水平每一个甲基化C碱基的甲基化水平均按如下公式进行计算:100*reads/total reads(例如:CpG位点的甲基化率=100*支持CG甲基化的reads/支持甲基化的reads+支持非甲基化的reads)全基因组平均甲基化水平反应了基因组甲基化图谱的总体特征。
Average methylation level for C, CG, CHG and CHH
2.5 全基因组甲基化数据分布趋势甲基化C碱基中CG, CHGG与CHH 的分布比例不同分布类型的甲基化C位点在不同物种基因组中出现比例不同,因此,各类型mC( mCG、mCHG和mCHH ) 的位点数目,及其在全部mC的位点中所
占的比例(例:mCHG所占比例= mCHG数目/mC的总数),在一定程度上反映了特定物种的全基因组甲基化图谱的特征。
不同分布类型甲基化 C 的数量及比例
此外,还统计如下数据:● CG、CHG和CHH中的所有C 的甲基化水平● 各个染色体中CG、CHG和CHH中C的甲基化水平● 统计不同基因区域内CG、CHG和CHH中C的甲基化水平● 不同基因元件区域中CG、CHG和CHH中C的甲基化水平● CHG、CHH中甲基化C附近的9bp序列的序列特征分析2.6 全基因组DNA甲基化图谱染色体水平的甲基化C 碱基的密度分布从染色体水平来描述甲基C碱基的的分布情况。蓝点表示以10kb的窗口统计甲基化C碱基的密度在染色体上的分布情况,光滑曲线则表示不同类型甲基化C碱基( G、CHG和CHH )的密度分布。
染色体上甲基化C密度分布
全基国组不同功能元件区域的甲基化水平分布
2.7差异性甲基化区域(DMR)分析在两个样品基因组相同位置上寻找包含5个CG的窗口,用CG甲基化水平的差异来寻找甲基化有差异的区域。确定每条染色体上有差异的区域长度和有差异的基因,并对这些基因做GO 聚类功能分
析,以分析差异相关基因是否有明显的功能聚类,即是否针对性地调控行使某类功能。DMR分析须基于两个样品进行差异比较。
DMR相关基因的GO聚类分析
3. 个性化信息分析根据客户的具体项目需求进行个性化分析。质控分离至受感染的E. coli, 是一段长488, 502bp 的未甲基化修饰的DNA片段。在Bisulfite处理中可以作为阴性对照用于计算Bisulfite处理的转化率。在文库构建过程中,λ-DNA会被加入到样品中(5ngλ-DNA/μg样品DNA),在完成测序后,通过信息分析来计算转化率。案例分析:研究者采用全基因组重亚硫酸盐测序方法,对小鼠胚胎干细胞(ES)和神经元祖细胞(NP)进行分析,构建小鼠单碱基对精度全基因组甲基化图谱。该张图谱显示,小鼠基因组中大部分区域(89.4%)呈现高甲基化状态,小部分区域(6.5%)呈现未甲基化状态,
另外还有一部分区域(4.1%)呈现出低甲基化状态(LMRs)。研究者对这小部分的低甲基化区域很感兴趣,对这些区域的特征进行详细的分析,他们发现转录因子以一种定向的方式促成LMRs模式出现。如果没有这些转录因子的作用,DNA依然保持甲基化和紧凑包装。由此研究者提出一种全新的表观遗传模式--转录因子介导的低甲基化调控模式。
小鼠胚胎肝细胞甲基化谱
基因组重测序
基因组重测序 背景介绍 全基因组重测序,是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。与已知序列比对,寻找单核苷酸多态性位点(SNP )、插入缺失位点(InDel ,Insertion/Deletion )、结构变异位点(SV ,Structure Variation )位点及拷贝数变化(CNV) 。 可以寻找到大量基因差异,实现遗传进化分析及重要性状候选基因的预测。涉 及临床医药研究、群体遗传学研究、关联分析、进化分析等众多应用领域。 随着测序成本的大幅度降低以及测序效率的数量级提升, 全基因组重测序已经成为研究人类疾病及动植物分子育种最为快速有效的方法之一。利用illumina Hiseq 2000 平台,将不同插入片段文库和双末端测序相结合,可以高效地挖掘基因序列差异和结构变异等信息, 为客户进行疾病研究、分子育种等提供准确依据。 重测序的两个条件:(1)该物种基因组序列已知;(2)所测序群体之间遗传性差异不大( >99% 相似度 ) 在已经完成的全基因组测序及其基因功能注释的基础上,采用全基因组鸟枪法(WGS )对DNA 插入片段进行双末端测序。 技术路线 生物信息学分析
送样要求 1.样品总量:每次样品制备需要大于5ug 的样品。为保证实验质量及延续性,请一次性提供至少20ug的样品。如需多次制备样品,按照制备次数计算样品总量。 2.样品纯度:OD值260/280应在1.8~2.0 之间;无蛋白质、RNA或肉眼可见杂质污染。 3.样品浓度:不低于50 ng/μL。 4.样品质量:基因组完整、无降解,电泳结果基因组DNA主带应在λ‐Hind III digest 最大条带23 Kb以上且主带清晰,无弥散。 5.样品保存:限选择干粉、酒精、TE buffer或超纯水一种,请在样品信息单中注明。 6.样品运输:样品请置于1.5 ml管中,做好标记,使用封口膜封好;基因组DNA如果用乙醇沉淀,可以常温运输;否则建议使用干冰或冰袋运输,并选择较快的运输方式。 提供结果 根据客户需求,提供不同深度的信息分析结果。
人类基因组重测序分析
6 首页 科技服务 医学检测 科学与技术 市场与支持 加入我们 关于我们提供领先的基因组学解决方案 Providing Advanced Genomic Solutions 诺禾致源 人类疾病基因组重测序分析图3 Circos 图 人类基因组重测序分析6项升级 Novo-Zhonghua Genomes 数据库注释 一些位点的突变可能在千人基因组中或在欧美人群中属于低频突变,但是对于中国人群来说却是常见突变。诺禾致源自建中国人数据库 Novo-Zhonghua Genomes,数据库中的所有样本均来自正常中国人群。已有研究表明,与国际通用的多人种数据库相比,使用单一人种数据库进行疾病研究,可以有效减少假阳性现象。 图2 真核生物基因的结构[6] 复杂疾病变异分类标准 DamLevel Variant Calling Variant Annotation Benign Likely Benign VUS Likely Pathogenic Custom knowledge Clinical Data Pathogenic Family Testing Published + in house data Population frequency Predictions: PolyPhen, SIFT, etc Amino acid conservation Published Disease Information Variant classification Candidate Variants Novo-Zhonghua Genomes 数据库注释 复杂疾病突变位点有害性分类 非编码区(Non-coding region)分析 疾病基因组 CNV/SV 分析 基于基因(Gene-based)的 Burden Analysis (复杂疾病散发样本) 可视化的数据结果展示 基于健康中国人群的千人测序数据,测序深度 > 30× 参考 ACMG 等,推出针对复杂疾病变异位点有害性的分类标准 应用 ENCODE 数据库最新内容,并结合国际通用数据库、自建数 复杂疾病突变位点有害性分类 基于美国医学遗传学会 ACMG[2]与 Duzkale H[3]提出的变异分类标准,诺禾致源疾病基因组信息分析团队推出了一套针对复杂疾病变异位点有害性的分类标准 DamLevel(如下图所示)。DamLevel 将变异位点的有害性分为5个层级:Pathogenic、Likely Pathogenic、VUS(Variant of uncertain significance)、Likely Begnin、Begnin,更好地鉴定个体遗传变异与疾病的相关性。 非编码区(Non-coding region)分析 基因组非编码区变异可以引发多种疾病,包括心脏类疾病、糖尿病、癌症、肥胖症等[4,5],但目前对非编码区突变的筛选和功能描述仍具挑战性。诺禾致源非编码区分析,应用 ENCODE 数据库最新内容对非编码区突变进行注释,通过国际通用数据库和自建的 Novo-Zhonghua Genomes 数据库进行频率筛选以及保守性过滤,精确定位非编码区中低频且保守的突变,筛选到与疾病相关的非编码区突变。 疾病基因组 CNV/SV 分析 CNV/SV 与基因表达、表型、人类疾病发生发展都有着非常密切的关系[7,8],诺禾致源疾病基因组信息分析团队研发了一整套 CNV/SV 筛选方法,包括有害性 CNV/SV 筛选和 de novo CNV/SV 分析(基于成三或成四家系)等。利用 DGV、DECIPHER、CNVD 等数据库对变异检出结果进行标记,从结果中进一步过滤掉良性 CNV/SV,经过一系列筛选后,准确鉴定个体 CNV/SV 遗传变异与疾病的相关性。 图4 CNV 分布图 表1 本次产品升级亮点 图5 Burden 分析结果的热图展示 1 2 3 4 5 Novo-Zhonghua Genomes 数据库注释 Novo-Zhonghua Genomes 数据库是诺禾致源自建针对 中国正常人群的数据库,助 力中国人群基因组信息解析。 复杂疾病突变位点 有害性分类 诺禾致源推出的复杂疾病变 异位点有害性的分类标准 (DamLevel),准确标识复杂 疾病的致病性突变位点。 非编码区 (Non-coding region)分析 应用 ENCODE 数据库最新内 容对非编码区进行注释、筛 选,精确定位非编码区中低 频且保守的突变。 疾病基因组 CNV/SV 分析 完整的有害性 CNV/SV 筛选 和 de novo CNV/SV 分析, 准确鉴定个体 CNV/SV 遗传 变异与疾病的相关性。 基于基因 (Gene-based)的 Burden Analysis 针对复杂疾病的研究,通过 检测疾病状态与基因变异的 相关性,寻找特定疾病(或 性状)的易感基因。 可视化的 数据结果展示 灵活易用的测序数据结果展 示,使大量复杂数据的分析 变得轻松而高效,提高数据 可读性。 ? log 10 ( P ? value ) Mutations of Genes Prioritized by Burden Analysis CIR1 PIGP CTSE PRB2 CYP HDAC1 GRK6 PIGK MYL6B EHD2 0810 246 Mutations 4 3 2 1 基于基因(Gene-based)的 Burden Analysis 关联分析是研究复杂疾病的1个重要方法,其通过检测疾病状态与基因变异的相关性,寻找特定疾病(或性状)的易感基因。通常是在具有不同表型的2组个体(一般为患病者和正常对照者)中,基于遗传位点(或基因、单体型)的频率分布差异,间接反映该遗传位点(或基因)可能与疾病(或性状)存在关联性。 Burden Analysis(Gene-based)基于复杂疾病的 case 和 control 散发样本,通过 Fisher's exact test 以及 SKAT 统计方法分析得到候选基因,针对候选基因可以进行富集分析(KEGG 富集分析和 GO 富集分析)与蛋白网络互作分析。 可视化的结果展示 诺禾致源疾病基因组信息分析团队,会为客户提供不断更新的变异注释、项目特异性分析和灵活易用的“变异-基因-疾病”可视化结果,让科学研究更轻松。 图6 疾病与基因关联性展示图 产品名称升级亮点 引领行 业新 标杆 参考文献 [1] Nagasaki M, Yasuda J, Katsuoka F, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals.[J]. Nature Communications, 2015, 6. 阅读原文 >> [2] Richards S, Aziz N, Bale S, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genetics in Medicine, 2015. 阅读原文 >> [3] Duzkale H, Shen J, McLaughlin H, et al. A systematic approach to assessing the clinical significance of genetic variants[J]. Clinical genetics, 2013, 84(5): 453-463. 阅读原文 >> [4] Yoshinari M, Akihiko M, Dongquan S, et al. A functional polymorphism in the 5' UTR of GDF5 is associated with susceptibility to osteoarthritis.[J]. Nature Genetics, 2007, 39(4):529-33. 阅读原文 >> [5] Kjong-Van L, Ting C. Exploring functional variant discovery in non-coding regions with SInBaD.[J]. Nucleic Acids Research, 2012, 41 (1):e7-e7. 阅读原文 >> [6] https://https://www.360docs.net/doc/de10234845.html,/wiki/Regulatory_sequence 阅读原文 >> [7] Sudmant P H, Rausch T, Gardner E J, et al. An integrated map of structural variation in 2,504 human genomes.[J]. Nature, 2015, 526 (7571):75-81. 阅读原文 >> [8] Birney E, Soranzo N. Human genomics: The end of the start for population sequencing.[J]. Nature, 2015, 526(7571):52-3. 阅读原文 >> 免费升级7-9月 新签合同 免费升级数据分析
全基因组重测序数据分析
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排 突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使 得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组 学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基 因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需 要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。
高通量基因组测序中 测序深度,覆盖度
高通量基因组测序中,什么是测序深度和覆盖度? 1G=1024M 测序深度是指测序得到的总碱基数与待测基因组大小的比值。假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M。(测序深度=总数据量20M/基因组大小2M=10X) 覆盖度是指测序获得的序列占整个基因组的比例。由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap。例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的。 1、全基因组重测序是对已知基因组序列的物种进行不同个体的基因 序的个体,通过序列比对,可以找到大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion/Deletion)、结构变异位点(SV, 技术路线 提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段(0.2~5Kb),加上接头, 进行cluster制备(Solexa)或E-PCR (SOLiD),最后利用Paired-End(Solexa)或者Mate-Pair(SOLiD)的方法对插入片段进行重测序。图1-1,以SOLiD为例,说明整个实验方案。
也称目标外显子组捕获,是指利用序列捕获技术将全基因组外显子区域DNA 捕捉并富集后进行高通量测序的基因组分析方法。是一种选择基因组的编码序列的高效策略,外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel 等具有较大的优势。 外显子(expressed region)是真核生物基因的一部分,它在剪接(Splicing)后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质。外显子是最后出现在成熟RNA中的基因序列,又称表达序列。既存在于最初的转录产物中,也存在于成熟的RNA分子中的核苷酸序列。在人类基因中大约有180,000外显子,占人类基因组的1%,约30MB。
群体进化-基于全基因组重测序
DNA样品总量: ≥3 μg 适用范围 样品要求 文库类型测序策略与深度 分析内容项目周期 群体进化(基于全基因组重测序) 标准分析时间为120天,个性化分析需根据项目实际情况进行评估 HiSeq PE150推荐测序深度≥5X/个体350 bp小片段DNA文库 1. 已有参考基因组序列的物种中不同亚群(自然群体) 2. 各亚群间划分明显,同一亚群内的个体有一定代表性 3. 每个亚群选取10个样本左右(推荐动物≥10个,植物≥15个) 4. 总体不少于30个样本与参考基因组比对群体SNP检测、注释及统计系统进化树构建群体遗传结构分析 群体主成分分析连锁不平衡分析选择消除分析候选基因GO和KEGG富集构建单体型图谱种群历史和有效群体大小 技术参数 针对已有参考基因组的物种,对其各亚种进行全基因组重测序获得基因组信息,通过与参考基因组比对,得到大量高准确性的SNP、InDel、SV等变异信息,讨论群体的遗传结构、遗传平衡和影响遗传平衡的因素,从而从分子层面揭示该物种的进化机制、环境适应性等系列问题。该技术能精准地得到全基因组内所有遗传信息,最大程度地挖掘出群体内遗传变异。诺禾具有丰富的群体遗传学项目经验,研究成果发表于Nature Genetics(Li, M, et al. 2013& Zhou, XM, et al. 2014)等。参考文献 [1] Li M, Tian S, Jin L, et al . Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars [J]. Nature genetics, 2013, 45(12): 1431-1438. [2] Zhan S, Zhang W, Niitepo ~ld K, et al . The genetics of monarch butterfly migration and warning colouration [J]. Nature, 2014.案例解析 [案例一] 家猪和藏猪的群体进化分析[1] 2013年,诺禾致源科技服务团队与四川农业大学研究者合作发表 该成果。本研究对6个代表性藏猪群体、5个四川盆地特有猪种, 共48个样本进行全基因组重测序,并结合55个欧亚野猪及家猪的 基因组数据进行群体遗传学分析。在藏猪中鉴定出低氧适应、能 量代谢等共268个适应高原环境的快速进化基因,揭示了藏猪高 原适应性的遗传机制。与自然选择相比,人工选择可更有效地塑 造驯养动物基因组;欧亚猪种存在明显的遗传背景差异,欧亚地 理隔离造成的遗传结构差异甚至超过了野生和驯化的差异。[案例二] 帝王蝶长距离迁飞遗传机制被解密[2] 北美地区的帝王蝶具有迁飞习性,而分布于热带地区的帝王蝶及 其近缘种不具有迁飞特性。该研究从涵盖当今世界上主要的帝王 蝶分布区域中,选取了包括迁飞型和非迁飞型的22个地理种群、 5个近缘种的101只班蝶属蝴蝶进行了全基因组重测序和群体遗传 学分析。结果表明,现存的帝王蝶起源于北美地区,且祖先属于 迁飞型,打破了先前认为包括鸟类等在内的迁飞物种均是热带起 源的普遍认知。其次,利用群体遗传学分析对全基因组进行精细 扫描发现,与飞行相关的肌肉发育进化是帝王蝶实现长距离迁飞 的主要适应性选择。 图1 藏猪及其它猪种的群体遗传结构 图2 帝王蝶样本分布及系统进化树
基因组重测序分析流程-代码文件
差异位点分析流程步骤分解 数据准备: mkdir 1.QC cd 1.QC ln -s /root/mdna-data/reseq/1.QC/*.fastq . Ls cd .. mkdir 2.mapping cd 2.mapping ln -s /root/mdna-data/reseq/2.mapping/ref.fasta . 步骤1:参考基因建索引 cd 2.mapping ##bwa建索引: bwa index ref.fasta Expected Result:得到一系列BWA 进行alignment 需要的文件。 ##samtools建索引: samtools faidx ref.fasta Expected Result:生成refgene.fasta.fai。每行都是fasta 文件中每条contig 的record,每条record 由contig name, size, location, basesPerLine 和bytesPerLine 组成。 ##生成字典: java -jar /root/mdna_software/picard-tools-1.102/CreateSequenceDictionary.jar R=ref.fasta O=ref.dict Expected Result:生成refgene.dict。描述fasta 文件内容,类似SAM header 格式。 步骤2:bwa比对 ##用bwa作比对: nohup bwa aln -e 3 -i 10 -t 1 -R 100 -q 20 ref.fasta ../1.QC/test_trim1.fastq -f 1.sai & nohup bwa aln -e 3 -i 10 -t 1 -R 100 -q 20 ref.fasta ../1.QC/test_trim2.fastq -f 2.sai & nohup bwa aln -e 3 -i 10 -t 1 -R 100 -q 20 ref.fasta ../1.QC/test_trim_unpaired.fastq -f s.sai & jobs
测序常用名词解释整理
高通量测序领域常用名词解释大全 什么是高通量测序? 高通量测序技术(,)是对传统测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术( , )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序( )。 什么是法测序(一代测序) 法测序利用一种聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(),并混入限量的一种不同的双脱氧核苷三磷酸()。由于缺乏延伸所需要的3基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。每一种和的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用光胶片放射自显影或非同位素标记进行检测。 什么是基因组重测序() 全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。 什么是测序
测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。获得一个物种的全基因组序列是加快对此物种了解的重要捷径。随着新一代测序技术的飞速发展,基因组测序所需的成本和时间较传统技术都大大降低,大规模基因组测序渐入佳境,基因组学研究也迎来新的发展契机和革命性突破。利用新一代高通量、高效率测序技术以及强大的生物信息分析能力,可以高效、低成本地测定并分析所有生物的基因组序列。 测序名词关系图 什么是
基于全基因组重测序获得的具LRR结构域基因的抗黄瓜白粉病功能鉴定
基于全基因组重测序获得的具LRR结构域基因的抗黄瓜白粉病 功能鉴定 黄瓜白粉病是黄瓜(CucumissativusL.)生产上的三大主要病害之一,发病时不但降低植株的光合效能,同时影响植株产量和果实品质,发病严重时常常引起30%左右的减产。黄瓜抗白粉病新品种选育及应用是克服白粉病危害的根本技术途径。 基于基因组测序技术和生物信息学的方法探究抗病基因已成为可能。本研究利用高通量Illumina测序技术,对实验室多年筛选获得的一个具有高抗白粉病且能稳定遗传的片段代换系SSL508-28和高感白粉病受体亲本D8进行了全基因组重测序,对比黄瓜9930参考基因组信息,在SSL508-28中发现了 468,616 个单核苷酸多态性位点(single nucleotide polymorphisms,SNPs)和 67,259小片段插入缺失位点(insertion/deletion,InDel),在D8 中获得了 537,352 个 SNPs 和 91,698个InDels。 通过对比SSL508-28与D8基因组,共得到了 15,682个SNPs和6,262个InDels,这些SNPs和InDels趋向于集中分布在五号染色体上。基于以上结果,我们对获得的SNPs和InDels进行了功能注释,发现有120个SNPs为非同义(non-synonymous)突变,30个InDels为移码突变(frameshift mutation),这些非同义突变SNPs和移码突变InDels分布在94个基因当中。 为了进一步验证94个突变基因对SSL508-28抗白粉病表型的贡献,我们对这94个基因进行了功能分类,其中有5个基因属于抗病(resistance,R)基因家族中NBS-LRR(Nucleotide binding site-leucine-rich repeats)类,利用 qRT-PCR 对这 5 个NBS-LRR基因在D8和SSL508-28中接种白粉菌前后的表达量进行检测,
重测序分析简介
重测序参考手册
目录 目录 (1) 1. 重测序简介 (3) 2. 重测序实验方法 (3) 基因组DNA抽提 (3) 基因组DNA样品建库 (3) 上机前定量 (4) 3. 重测序分析内容 (4) 重测序分析流程 (5) 重测序分析内容 (5) 4. 重测序重要技术参数 (6) 5. 重测序分析内容解释 (6) 6. 重测序分析内容示例 (6) SNP、INDEL的样本差异分析 (12) 7. 成功分析案例/或已发表论文 (14) 8. 概念及常用工具链接 (14)
1. 重测序简介 全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。全基因组重测序的个体,通过序列比对,可以找到大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion/Deletion)、结构变异位点(SV,Structure Variation)位点。众信可以协助客户,通过生物信息手段,分析不同个体基因组间的结构差异,同时完成注释。 2. 重测序实验方法 提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段(0.2~5Kb),加上接头, 进行cluster制备(Solexa)或E-PCR (SOLiD),最后利用Paired-End或者Mate-Pair的方法对插入片段进行重测序。 实验步骤主要包括以下几点: 基因组DNA抽提 不同生物(植物、动物、微生物)的基因组DNA的提取方法有所不同; 不同种类或同一种类的不同组织因其细胞结构及所含的成分不同,分离方法也有差异。在提取某种特殊组织的DNA时必须参照文献和经验建立相应的提取方法, 以获得可用的DNA大分子。尤其是组织中的多糖和酶类物质对随后的酶切、PCR反应等有较强的抑制作用,因此用富含这类物质的材料提取基因组DNA时, 应考虑除去多糖和酚类物质。 基因组DNA样品建库 这是样品准备过程中最主要的环节,也就是真正意义上的建库(通常我们所说的建库包括整个样品准备的过程)。 样品片段化(Covaris) Covaris利用超声波剪切DNA,并将传统超声波法可控制化、精确化。DNA可以在小体积中被剪切,减少了因为蒸发带来的样品损耗,并且被剪切的DNA片段大小之间的偏差较小。Covaris剪切的片段大小较小,并且片段大小范围较传统超声波法窄。选择合适的打断参数条件,使最后打断的DNA片段大小集中在300-500bp范围内。 末端修复 使用Covaris剪切的DNA片段都会形成一些杂合的末端,其中包括了3’ 端悬垂结构、
全基因组重测序解决方案
全基因组重测序解决方案 ※ 概述 全基因组重测序是对已有参考序列(Reference Sequence)的物种的不同个体进行基因组测序,并以此 为基础进行个体或群体水平的差异性分析。通过全基因组重测序,研究者可以找到大量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插入缺失(InDel,Insertion/Deletion)、结构变异(Structure Variation,SV)等变异位点。这在人类疾病及动植物育种研究等方面具有重大的指导意义。基 于此,我们推出重测序的完整解决方案,协助您一起探索生物奥秘。 ※ 实验技术流程 ※ 生物信息分析策略
1、数据量产出分析 总碱基数量、Totally mapped reads、Uniquely mapped reads统计,测序深度分析。 2、一致性序列组装 与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基 位点的最大可能性基因型,并组装出该个体基因组的一致序列。 3、SNV检测及在基因组中的分布 提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到 可信度高的SNV数据集。并根据参考基因组序列对检测到的变异进行注释。 4、InDel检测及在基因组的分布 在进行mapping的过程中,进行容Gap的比对并检测可信的Short InDel。在检测过程中,Gap的长度为 1~5个碱基。 5、Structure Variation检测及在基因组中的分布 检测主要的结构变异类型有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比 对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。 ※ 参考文献 1、Erin D, et al., A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature, 2010, 463:184-190. 2、Carl-Johan Rubin, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature, 2010, 464:587-597. 3、Qingyou Xia,et al. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science, 2009, 326:433-436.
重测序-全基因组选择(GS)
首页 科技服务 测序指南 基因课堂 市场活动与进展 文章成果 关于我们 全基因组选择1. Meuwissen T H, Hayes B J, Goddard M E.Prediction of total genetic value using genome-wide dense marker maps[J]. Genetics, 2001, 157(4): 1819 1829. 阅读原文>> 2. Haberland A M, Pimentel E C G, Ytournel F, et al. Interplay between heritability, genetic correlation and economic weighting in a selection index with and without genomic information[J]. Journal of Animal Breeding and Genetics, 2013, 130(6): 456-467. 阅读原文>> 3. Wu X, Lund M S, Sun D, et al. Impact of relationships between test and training animals and among training animals on reliability of genomic prediction[J]. Journal of Animal Breeding and Genetics, 2015, 132(5): 366-375. 阅读原文>> 4. Goddard M E ,Hayes BJ. Genomic selection [J]. Journal of Animal Breeding and Genetics,2007,124:323:330. 阅读原文>> 5. Heffner E L, Sorrells M E, Jannink J L. Genomic selection for crop improvement [J]. Crop Science, 2009, 49(1): 1-12. 阅读原文>> 参考文献 全基因组选择简介 Meuwissen等[1]在2001年首次提出了基因组选择理论(Genomic selection , GS),即利用具有表型和基因型的个体来预测只具有基因型不具有表型值动植物的基因组育种值(GEBV)。 例如,提高奶牛的产奶量一直是奶牛研究者的研究重点,传统育种的方法需要牛生长至成年后,才能进行产奶量的测定,再进行后续的育种进程。如果在犊牛刚出生时就可以通过某种技术预测出其产奶量,就可以大大的减少育种时间,节省大量的育种成本。 全基因组选择(GS)利用覆盖全基因组的高密度分子遗传标记进行标记辅助选择,可以在奶牛的幼年时期就预测出其生产性状和营养性状,快速筛选出具有优良性状的奶牛或者种公牛,加速育种的进程。 全基因组选择技术参数 提供领先的基因组学解决方案 Leading Edge Genomic Services & Solutions 动植物重测序变异检测BSA性状定位遗传图谱群体进化全基因组关联分析Hi-C测序 人类基因组测序全基因组测序外显子测序目标区域测序单细胞基因组测序 动植物基因组测序全基因组survey 全基因组 de novo 测序泛基因组测序组装变异检测 微生物基因组测序16S/18S/ITS等扩增子测序细菌基因组 de novo 测序真菌基因组 de novo 测序微生物重测序宏基因组测序 建库测序建库测序 诺禾致源微信文章精彩阅读 >> 版权所有:北京诺禾致源科技股份有限公司 转录调控测序 真核有参转录组测序医学转录组测序真核无参转录组测序比较转录组与泛转录组测序原核转录组测序宏转录组测序单细胞转录组测序LncRNA测序circRNA测序small RNA测序ChiP-seq RIP-seq 全基因组甲基化测序 GS 重测序新产品发布 群体大小 参考群体的选择十分重要,表型信息及固定效应信息记录需要准确完整。此外,选择出 的参考群体要满足内部亲缘关系比较远,数量达到1000个以上[2]。候选群体最好与参考群体的亲缘关系较近,这样可以保证育种值预测的准确性[3]。 测序策略 测序深度:平均每个样本≥10×;测序平台:Illumina HiSeq PE150测序; 全基因组选择技术优势 全基因组选择与传统的分子标记辅助选择相比,具有很多优势[5]: 能够在得到物种个体DNA的时候即对其进行育种值评估,可以缩短世代间隔,加快遗传进展并且降低经济投入。 全基因组范围内的标记能够解释尽可能多的遗传变异,可以对遗传效应进行较为准确的检测和估计。 能够较准确的评估遗传力较低、难测定的性状或测定费用较高的性状。 通过基因组选择的方式,即使单个标记的效应很微小,导致遗传变异的所有遗传效应也都能够被SNP标记捕获, 所以比传统的基于系谱和表型数据的最佳线性无偏模型得到更高的可靠性。 a b c d
全基因组重测序数据分析
全基 1. 简 通过变(d 的功况,dise 比较 实验 (1)(2) 基因组重测序简介(Introduc 过高通量测序识deletioin, du 功能性进行综合杂合性缺失ease (cance 较基因组学,群验设计与样本 Case-Contr )家庭成员组序数据分析 ction) 识别发现de plication 以及合分析;我们(LOH )以及r )genome 中群体遗传学综ol 对照组设计 组设计:父母novo 的som 及copy numb 们将分析基因及进化选择与中的mutation 综合层面上深计 ; -子女组(4 人matic 和germ ber variation 因功能(包括与mutation 之n 产生对应的深入探索疾病基人、3 人组或m line 突变,)以及SNP miRNA ),重之间的关系;以的易感机制和基因组和癌症多人); 结构变异-SN 的座位;针对重组率(Rec 以及这些关系功能。我们将症基因组。 NV ,包括重排对重排突变和combination )系将怎样使得 将在基因组学排突 SNP )情在 学以及
初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。 高级数据分析 1.测序短序列匹配(Read Mapping) (1)屏蔽掉Y染色体上假体染色体区域(pseudo-autosomal region), 将Read与参考序列NCBI36进行匹配(包括所有染色体,未定位的contig,以及线粒体序列mtDNA(将用校正的剑桥参考序列做替代))。采用标准序列匹配处理对原始序列文件进行基因组匹配, 将Read与参考基因组进行初始匹配;给出匹配的平均质量得分分布; (2)碱基质量得分的校准。我们采用碱基质量校准算法对每个Read中每个碱基的质量进行评分,并校准一些显著性误差,包括来自测序循环和双核苷酸结构导致的误差。 (3)测序误差率估计。 pseudoautosomal contigs,short repeat regions(包括segmental duplication,simple repeat sequence-通过tandem repeat识别算法识别)将被过滤; 2. SNP Calling 计算(SNP Calling) 我们可以采用整合多种SNP探测算法的结果,综合地,更准确地识别出SNP。通过对多种算法各自识别的SNP进行一致性分析,保留具有高度一致性的SNP作为最终SNP结果。这些具有高度一致性的SNP同时具有非常高的可信度。在分析中使用到的SNP识别算法包括基于贝叶斯和基因型似然值计算的方法,以及使用连锁不平衡LD或推断技术用于优化SNP识别检出的准确性。 统计SNV的等位基因频率在全基因组上的分布
重测序
全基因组重测序 项目简介 全基因组重测序是对已有参考序列(Reference Sequence)的物种的不同个体进行基因组测序,并以此为基础进行个体或群体水平的差异性分析。通过这种方法,可以寻找出大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion Deletion),结构变异位点(SV,Structure Variation),拷贝数变异(Copy Number Variation,CNV)等变异信息,从而获得生物群体的遗传特征。这对在群体水平上研究物种的进化历史、环境适应性、自然选择等方面具有重大意义。利用全基因组重测序有助于快速发现与动植物重要性状相关的遗传变异,缩短分子育种的实验周期;有助于发现人类疾病相关的重要变异基因,加快生物医药研发的速度等,这对人类疾病及动植物育种研究等方面具有重大的指导意义。 技术流程 提取基因组DNA后,采用物理方法随机打断,选择性回收所需长度的DNA片段(0.2~5Kb),并在两端连接接头以构建测序文库,进行桥式PCR(Bridge Amplification)制备Cluster,最后利用Paired-End的方法对插入片段进行重测序。 生物信息分析 1.数据量产出 总碱基数量、Totally mapped reads、Uniquely mapped reads统计,测序深度分析。 2.一致性序列组装 与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。 3.SNP检测及在基因组中的分布 提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基因组序列对检测到的变异进行注释。 4.InDel检测及在基因组的分布 在进行mapping的过程中,进行容Gap的比对并检测可信的Short InDel。在检测过程中,Gap的长度为1~5个碱基。 5.Structure Variation检测及在基因组中的分布
基因组测序术语解释
DNA关键词: WG-BSA (全基因组重测序BSA) 对已有参考基因组序列的物种的所有作图群体(F1、F2、RIL、DH 和BC1等),对亲本进行个体重测序,对某个极端性状材料混池测序,检测SNP,获得与性状紧密关联的分子标记和精细定位区域,是目前最高效的基因定位方法。通过选取某个极端性状,利用高效率低成本的混池测序技术,勿需开发分子标记进行遗传图的构建,快速定位与性状相关的候选QTL。 MP-Reseq (多混池全基因组重测序) 针对特有的优良地方品种中的不同品种/品系,通过群体内pooling 建库的方法,进行全基因组重测序,采用生物信息学方法全基因组范围内扫描变异位点,能快速的定位不同混池样品基因组中明显经过人工或自然选择的区域,检测与性状相关的基因区域及其功能基因。 全基因组个体重测序 基于全基因组重测序的变异图谱通过测序手段结合生物信息分析研究同一物种不同个体之间的变异情况,获得大量的变异信息,如SNP、Indel、SV 等。主要可以快速地获得大量的分子标记以及不同个体在基因组水平上的差异。 全基因组关联分析-GWAS 通过重测序对动植物重要种质资源进行全基因组基因型鉴定,与关注的表型数据进行全基因组关联分析,找出与关注表型相关的SNP位点,定位数量性状基因,与数量性状相关的基因紧密连锁的SNP标记,后续可用于分子标记辅助育种,助力育种进程。 全基因组重测序-遗传进化 通过对来自全国各地、具有代表性的XX 份XX 材料进行全基因组重测序,检测SNP、Indel、SV,并利用获得的SNP 与SV 数据进行群体多样性分析,包括连锁不平衡分析、群体进化分析、群体结构分析、群体主成分分析等。 全基因组重测序-遗传图谱 基于全基因组重测序技术对已有参考基因组序列的物种进行个体或群体的全基因组测序,利用高性能计算平台和生物信息学方法,检测单核苷酸多态性位点(SNP),并计算多态性标记间的遗传连锁距离,绘制高密度的遗传图谱。通过与表型性状进行关联分析,利用获得的强关联性标记进行下游基因的精细定位。遗传图可用于分子标记辅助育种,重要性状候选基因克隆,辅助基因组组装,比较基因组学等研究。 细菌基因组de novo 测序 细菌是生物的主要类群之一,是所有生物中数量最多的一类。细菌广泛分布于土壤和水中,或者与其他生物共生,也有部分种类分布在极端环境中,例如温泉,甚至是放射性废弃物中。由于细菌自身的营