DNA序列比对同源性分析图解BLAST
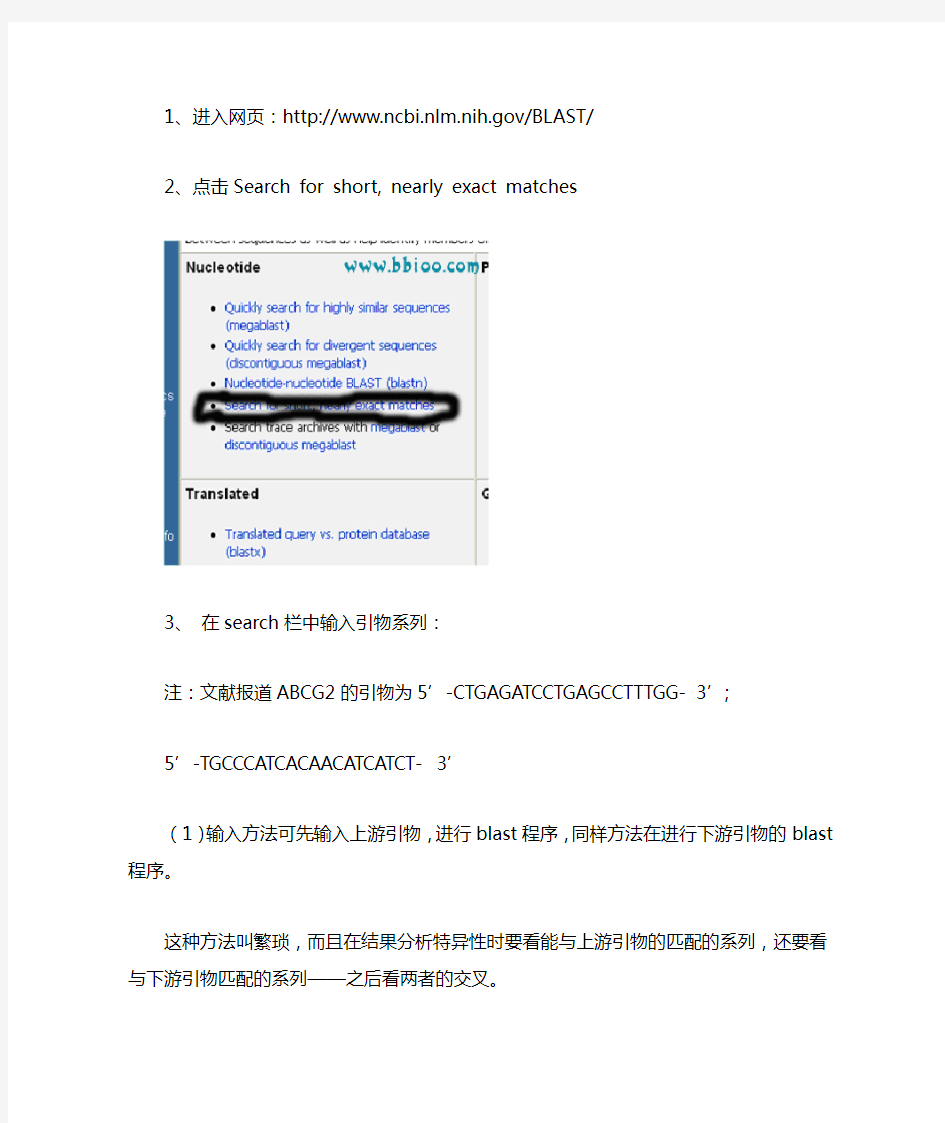
1、进入网页:https://www.360docs.net/doc/1315186654.html,/BLAST/
2、点击Search for short, nearly exact matches
3、在search栏中输入引物系列:
注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;
5’-TGCCCATCACAACATCATCT-3’
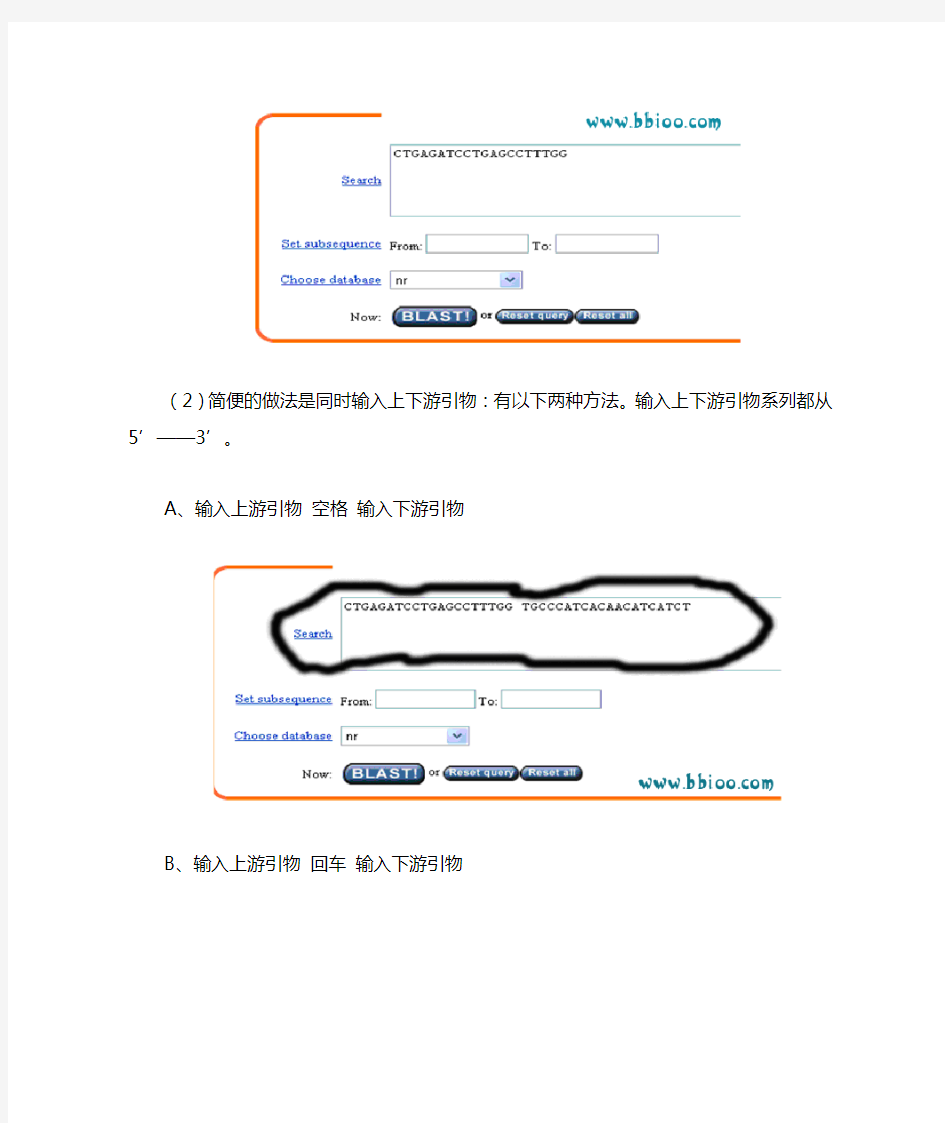
(1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。
这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。输入上下游引物系列都从5’——3’。
A、输入上游引物空格输入下游引物
B、输入上游引物回车输入下游引物
4、在options for advanced blasting中:
select from 栏通过菜单选择Homo sapiens
Expect后面的数字改为10
5、在format中:
select from 栏通过菜单选择Homo sapiens Expect后面的数字填上0 10
6、点击网页中最下面的“BLAST!”
7、出现新的网页,点击Format!
8、等待若干秒之后,出现results of BLAST的网页。该网页用三种形式来显示blast的结果。(1)图形格式:
图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分
图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补
图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配
通过点击相应的bar可以得到匹配情况的详细信息。
(2)结果信息概要:
从左到右分别为:
A、数据库系列的身份证:点击之后可以获得该序列的信息
B、系列的简单描述
C、高比值片段对(high-scoring segment pairs, HSP)的字符得分。按照得分的高低由大到小排列。得分的计算公式=匹配的碱基×2+0.1。举例:如果有20个碱基匹配,则其得分为40.1。
D、E值:代表被比对的两个序列不相关的可能性。。E值最低的最有意义,也就是说序列的相似性最大。设定的E值是我们限定的上限,E值太高的就不显示了
E、最后一栏有的有UEG的字样,其中:
U代表:Unigene数据库
E代表:GEO profiles数据库
G代表:Gene数据库
(3)结果详细信息:
①圈出来的部分代表序列的信息
②第一个大括号代表上游引物与该序列的正链的匹配情况:
共有21个碱基匹配,得分42.1分,E值为0.020
上游引物与序列的2143~2163位点匹配
③第二个大括号代表下游引物与该序列的负链的匹配情况:
共有20个碱基匹配,得分40.1分,E值为0.077。
下游引物与该序列的29360~29379位点互补
注意点:
①上游引物为20个碱基,为什么会变成21个碱基呢?这是因为下游引物的第一个碱基为T,刚好与系列的2163位点的T匹配,因此下游引物的开头的第一个碱基被当成了上游引物了。同理,上游引物的最后一个碱基为G,被当成了下游引物了。通过寻找有没有与1~20位点、20~40位点完全匹配的序列,就可以避免这个因素的干扰了。
②为什么与上下游引物匹配的ABCG2序列有多种?
A、为同一个基因来源的不同的mRNA片段
B、为该基因的DNA系列
C、为同一个基因来源的不同的cDNA片段。
结果判断:
①验证文献报道的引物是否正确:如果你可以在所显示的结果中找出你的目的基因,一般说明你的引物正确性没问题。如果你blast后没有发现你的目的基因,或者分值很低,该引物就可能不适合用
②检测该对引物是否可与其它序列匹配,引起PCR的非特异性扩增。如果找到了你的目的基因名称,而且找到了一大批同物种的不同基因,(上下游引物分别搜索到相同的基因),而且分数也较高。这时表明你的引物设计的特异性不高,极有可能在你的扩增产物中出现非特异性产物。
临床微生物检测的基因同源性分析
临床微生物检测的基因同源性分析 医院感染的流行病学研究是临床微生物学工作者的重要课题,在医院感染监测的研究中,一个很重要的问题就是如何确定感染途径和传播途径,以采取有效预防和控制措施,从而防止医院感染或者暴发流行,因此对微生物鉴定和菌株同源性分析提出了更高的要求。 目前,传统的细菌鉴定和同源性分析技术已不能很好地满足医院感染诊断和流行病学调查的需要,从型、亚型、株,甚至分子水平上去认识细菌变得愈来愈重要了。近年来分子生物学的理论和技术在细菌感染诊断中的渗透和广泛应用,使得细菌鉴定、耐药基因的检测、分子流行病学调查变得更加准确、简洁和快速。细菌DNA同源性分析技术如脉冲场凝胶电泳、聚合酶链反应、DNA探针杂交以及序列分析等方法是目前在分子水平上分析细菌的主要手段,它在鉴定细菌感染的爆发、确定院内交叉感染、感染病原菌之间的基因同源性等方面有着重要的作用,本文将对常用基因同源性分析方法的机理和过程做简要综述。 一、质粒分型(Plasmid profile assay): 1、原理: 质粒是可移动的染色体外元件,可以自发丢失或者被宿主稳定地获得,因此流行病学上相关的分离菌株有可能表现出不同的质粒图谱。把质粒提取出来,进行常规的琼脂糖电泳分析,就可以知道分离菌株携带质粒的大小和数目。 2、实验过程:a.质粒抽提;b.0.8%琼脂糖电泳;c.EB染色、凝胶成像。 3、实验方法的评价: 优势: a.步骤比较简单,对实验仪器要求不高。 b.在评价那些从局限的时间和地点(如一个医院中的急性爆发)分离出来的菌株非常有效。 缺点: a.实验结果的重复性不好。 b.分辨力不高。 二、染色体DNA的限制性内切核酸酶分析 (Restriction Endonuclease Assay REA) 1、原理: 限制性内切核酸酶(REA)在特异的核酸识别序列切割DNA,DNA被消化后,所得到的限制性片段的数目和大小是由酶的识别位点和DNA的组成共同决定的。在传统的限制性内切核酸酶分析中,人们用含有相对多的限制性位点的内切核酸酶消化细菌的DNA,这样就会得到成百上千条长度在0.5-50Kb范围内的DNA 片段。恒定的电场琼脂糖电泳,可以根据分子质量的大小将这些DNA片段分开,然后通过EB染色并在紫外灯下观察其图谱。同一种的不同分离菌株的DNA序列的变异可造成限制性位点数目和分布的变化,所以可以导致其REA图谱出现差异。 2、实验流程:a.染色体DNA抽提;b.限制性内切酶消化DNA(主要是Hind III); c.0.7%琼脂糖电泳; d.EB 染色、凝胶成像。
DNA序列比对同源性分析图解BLAST
1、进入网页:https://www.360docs.net/doc/1315186654.html,/BLAST/ 2、点击Search for short, nearly exact matches 3、在search栏中输入引物系列: 注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’; 5’-TGCCCATCACAACATCATCT-3’ (1)输入方法可先输入上游引物,进行blast程序,同样方法在进行下游引物的blast程序。 这种方法叫繁琐,而且在结果分析特异性时要看能与上游引物的匹配的系列,还要看与下游引物匹配的系列——之后看两者的交叉。
(2)简便的做法是同时输入上下游引物:有以下两种方法。输入上下游引物系列都从5’——3’。 A、输入上游引物空格输入下游引物 B、输入上游引物回车输入下游引物 4、在options for advanced blasting中: select from 栏通过菜单选择Homo sapiens Expect后面的数字改为10
5、在format中: select from 栏通过菜单选择Homo sapiens Expect后面的数字填上0 10
6、点击网页中最下面的“BLAST!” 7、出现新的网页,点击Format!
8、等待若干秒之后,出现results of BLAST的网页。该网页用三种形式来显示blast的结果。(1)图形格式: 图中①代表这些序列与上游引物匹配、并与下游引物互补的得分值都位于40~50分 图中②代表这些序列与上游引物匹配的得分值位于40~50分,而与下游引物不互补 图中③代表这些序列与下游引物互补的得分值小于40分,而与上游引物不匹配 通过点击相应的bar可以得到匹配情况的详细信息。
同源性分析标准操作规程
同源性分析标准操作规程 持有部门:检验科 制定部门:丰都县人民医院医院感染管理科执行时间: 2013年11月1日 一、同源性分析的应用 包括感染暴发的判断,感染病原菌的确定及感染源的寻找。 二、基本方法 1.细菌的表型特征分型技术(如血清型、耐药表型等)。 2.基因分型技术(如.PFGE、Rep—PCR、AFLP等)。 三、操作步骤 (一)表型分型(血清型、耐药表型) 1.标本孵育:从患者感染部位采集标本,并接种于相应培养基(培养瓶)中孵育。 2.分离菌种:分离病原菌(细菌或真菌),并鉴定细菌(真菌)种类。 3.血清分型:如可能则对同种细菌进行血清分型,如军团菌等。 4.药敏试验:根据细菌(真菌)种类选择药敏卡(纸片),进行药敏试验。 5.结果分析:分析血清型和药敏谱,如结果相同或药敏相差不大则提示有同源性。 (二)基因分型(PFGE) 1.菌栓制备:将细菌悬液与2%低熔点琼脂糖凝胶混合,制备菌栓。 2.细菌消化:分别用含溶菌酶和(或)蛋白酶K的裂解液对菌栓进行消化。 3.洗涤菌栓:可用无菌水反复清洗或PMSF中和多余的蛋白酶K。 4.酶切:按照各种不同限制性内切酶的说明进行酶切。 5.制胶:用PF(讵电泳凝胶模具制备:PF(逼级琼脂糖凝胶。 6.电泳:根据不同细菌酶切片段的大小选择适当的脉冲参数,进行电泳。 7.凝胶成像:胶块染色后在凝胶成像仪下成像。 8.结果分析:根据Tenover等制定的标准对凝胶图像进行分析,判断菌株之间的同源性。 四、注意事项 1.不同的分型方法,在分型能力、重复性、分辨能力、操作和成本等方面都不尽相同,可根据情况进行选择。 2.表型分型方法的分辨能力普遍比基因分型方法低,但简便、快速,适用于对感染暴发的初筛。
小麦TaeEF1β基因的克隆、同源性及表达分析
浙江农业学报ActaAgriculturaeZhejiangensis?2019?31(1):98-103http://www.zjnyxb.cn陈炫?张天烨?羊健?等.小麦TaeEF1β基因的克隆二同源性及表达分析[J].浙江农业学报?2019?31(1):98-103. DOI:10 3969/j.issn.1004 ̄1524 2019 01 13 收稿日期:2018 ̄03 ̄25 基金项目:国家自然科学基金(3150160)?国家农业产业体系(CARS ̄3 ̄1)?国家小麦转基因专项(2016ZX08002001) 作者简介:陈炫(1992 )?女?陕西咸阳人?硕士研究生?主要从事植物病理学研究?E ̄mail:vivianccx@163.com?通信作者?陈剑平?E ̄mail:jpchen2001@126.com小麦TaeEF1β基因的克隆二同源性及表达分析 陈一炫1?张天烨1?羊一健2?张恒木2?陈剑平2?? (1.浙江农林大学林业与生物技术学院?浙江杭州311300?2.浙江省农业科学院病毒学与生物技术研究所?浙江杭州310021) 摘一要:真核翻译延伸因子(eukaryotictranslationelongationfactor?eEFs)是一种重要的多功能调控蛋白?eEF1β是eEF1的组成部分?在蛋白质生物合成过程中发挥着重要的作用?本文通过RT ̄PCR扩增克隆小麦(TriticumaestivumL.)的eEF1β基因?并命名为TaeEF1β?氨基酸同源性分析发现?TaeEF1β具有高度保守性?且其保守结构域位于137~226aa处?qRT ̄PCR结果表明?中国小麦花叶病毒(Chinesewheatmosaicvi ̄rus?CWMV)侵染小麦植株后?可以诱导TaeEF1β基因转录水平的上调表达?另外?本文也进一步分析了TaeEF1β基因在小麦根二茎二叶的表达水平和CWMV侵染不同时间点的表达情况? 关键词:真核翻译延伸因子?中国小麦花叶病毒?同源性分析 中图分类号:S435 12文献标志码:A文章编号:1004 ̄1524(2019)01 ̄0098 ̄06Genecloning?homologyandexpressionanalysisofTaeEF1βinTriticumaestivumL.CHENXuan1?ZHANGTianye1?YANGJian2?ZHANGHengmu2?CHENJianping2?? (1.CollegeofForestryandBiotechnology?ZhejiangA&FUniversity?Hangzhou311300?China?2.InstituteofVirolo ̄gyandBiotechnology?ZhejiangAcademyofAgriculturalSciences?Hangzhou310021?China) Abstract:Eukaryotictranslationelongationfactor(eEFs)isanimportantmultifunctionalprotein.eEF1βwassub ̄unitoftheeukaryotictranslationelongationfactor ̄1(eEFs ̄1)complexandplayedanimportantroleinproteinbio ̄synthesis.TheeEF1βgenewasobtainedbycloningwithRT ̄PCRfromwheatandnamedasTaeEF1β.TheaminoacidsequencesofTaeEF1βwerehighlyconservedandtheconserveddomainwaslocatedin137 ̄226aa.TheresultsofqRT ̄PCRshowedthatTaeEF1βwereup ̄regulatedintheCWMV ̄infectedplants.Inthisstudy?theexpressionofTaeEF1βinthestems?leavesandrootsofwheatanddifferentstagesofCWMVinfectionwasalsodetectedbyqRT ̄PCR.Keywords:eukaryotictranslationelongationfactor?Chinesewheatmosaicvirus?homologyanalysis 一一真核翻译延伸因子(eukaryotictranslatione ̄longationfactor?eEFs)最早作为一个对噬菌体Qβ 的依赖于RNA的RNA聚合酶(RNA ̄dependentRNApolymerase?RdRp)活性具有重要作用的辅助因子被发现并鉴定[1]?在真核细胞中包括3类延伸因子?分别为eEF1α(eukaryotictranslationelongationfactor1α)二eEF1β和eEF2[2]?EF1β在真核生物中的结构比在细菌中更复杂?该蛋白由EF1β ̄alpha二EF1β ̄gamma和EF1β ̄beta三个亚基组成?在蛋白质的合成过程中?eEF1β主要作为
BioEdit及MEGA分析序列同源性简介
利用系统进化分析软件对序列进行同源性分析 1.0目的 1.1为了保持国际上各个耐药性实验室的高检验水准,进行分子进化分 析是有很重要作用的。它不仅可以保证流行病学目的顺利实现,而 且有利于发现检测阶段可能产生的潜在的交叉污染。 1.2利用此软件进行分析所得的信息对于进行艾滋病毒在人群中传播 的流行病学研究具有十分重要的意义。 1.3通过对实验室之前分析的数据与当前数据进行比较,可以发现在此 前实验过程中由于标本处理不当所导致的潜在的实验室污染。 1.4对于确保得到高质量的实验结果并及时发现可能出现在实验室里 的问题具有重要意义。 2.0仪器设备 2.1计算机一台。 2.2Windows 95以上的操作系统。 2.3BioEdit 以及MEGA 4 分析软件。 2.3.1均为免费软件,可以从互联网上下载,在计算机上进行安装。 3.0操作过程 3.1局限及要求 3.1.1经过序列编辑软件拼接处理后的txt文件或fasta文件均可, 例如ChromasPro软件。 3.1.2可以在MEGA上分析的分子序列或距离矩阵数据。 3.1.3Mega 4软件只能将长度相等的序列转换为MEGA输出文 件,因此,任何多序列文件必须通过BioEdit软件进行对齐 修剪,然后才能进入通过Mega软件转换成*.meg格式进行 分析。 3.1.4该数据集的大小受限于计算机上可用的物理(RAM)和虚 拟内存。 3.1.5分析的序列必须包括两个或两个以上长度相同的序列,所有 序列分析之前必须用MEGA软件对齐。 3.1.6核苷酸和氨基酸序列应该用英文字母连续书写,不区分大小 写。一些特殊的特殊符号,例如表示对齐缺口,碱基缺失等 的符号也可以包含在序列中。 3.1.7空格和制表符经常用于数据文件中,因此会被MEGA忽略。 ASCII字符,如(.)(- )(?),一般都作为特殊符号
临床微生物检测的基因同源性分析
临床微生物检测的基因 同源性分析 Revised as of 23 November 2020
临床微生物检测的基因同源性分析 医院感染的流行病学研究是临床微生物学工作者的重要课题,在医院感染监测的研究中,一个很重要的问题就是如何确定感染途径和传播途径,以采取有效预防和控制措施,从而防止医院感染或者暴发流行,因此对微生物鉴定和菌株同源性分析提出了更高的要求。 目前,传统的细菌鉴定和同源性分析技术已不能很好地满足医院感染诊断和流行病学调查的需要,从型、亚型、株,甚至分子水平上去认识细菌变得愈来愈重要了。近年来分子生物学的理论和技术在细菌感染诊断中的渗透和广泛应用,使得细菌鉴定、耐药基因的检测、分子流行病学调查变得更加准确、简洁和快速。细菌DNA同源性分析技术如脉冲场凝胶电泳、聚合酶链反应、DNA 探针杂交以及序列分析等方法是目前在分子水平上分析细菌的主要手段,它在鉴定细菌感染的爆发、确定院内交叉感染、感染病原菌之间的基因同源性等方面有着重要的作用,本文将对常用基因同源性分析方法的机理和过程做简要综述。 一、质粒分型(Plasmidprofileassay): 1、原理: 质粒是可移动的染色体外元件,可以自发丢失或者被宿主稳定地获得,因此流行病学上相关的分离菌株有可能表现出不同的质粒图谱。把质粒提取出来,进行常规的琼脂糖电泳分析,就可以知道分离菌株携带质粒的大小和数目。
3、实验方法的评价: 优势: a.步骤比较简单,对实验仪器要求不高。 b.在评价那些从局限的时间和地点(如一个医院中的急性爆发)分离出来的菌株非常有效。 缺点: a.实验结果的重复性不好。 b.分辨力不高。 二、染色体DNA的限制性内切核酸酶分析(RestrictionEndonucleaseAssayREA) 1、原理: 限制性内切核酸酶(REA)在特异的核酸识别序列切割DNA,DNA被消化后,所得到的限制性片段的数目和大小是由酶的识别位点和DNA的组成共同决定的。在传统的限制性内切核酸酶分析中,人们用含有相对多的限制性位点的内切核酸酶消化细菌的DNA,这样就会得到成百上千条长度在范围内的DNA片段。恒定的电场琼脂糖电泳,可以根据分子质量的大小将这些DNA片段分开,然后通过EB染色并在紫外灯下观察其图谱。同一种的不同分离菌株
多重序列比对及系统发生树的构建
多重序列比对及系统发生树的构建 作者:佚名来源:生物秀时间:2007-12-31 【实验目的】 1、熟悉构建分子系统发生树的基本过程,获得使用不同建树方法、建树材料和建树参数对建树结果影响的正确认识; 2、掌握使用Clustalx进行序列多重比对的操作方法; 3、掌握使用Phylip软件构建系统发生树的操作方法。 【实验原理】 在现代分子进化研究中,根据现有生物基因或物种多样性来重建生物的进化史是一个非常重要的问题。一个可靠的系统发生的推断,将揭示出有关生物进化过程的顺序,有助于我们了解生物进化的历史和进化机制。 对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行比对(alignment)。⑵要构建一个进化树(phyligenetic tree)。构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离。独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。⑶对进化树进行评估,主要采用Bootstraping法。进化树的构建是一个统计学问题,我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。模拟的进化树需要一种数学方法来对其进行评估。不同的算法有不同的适用目标。一般来说,最大简约性法适用于符合以下条件的多序列:i 所要比较的序列的碱基差别小,ii 对于序列上的每一个碱基有近似相等的变异率,iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多(大于几千个碱基);
多序列比对
在寻找基因和致力于发现新蛋白的努力中,人们习惯于把新的序列同已知功能的蛋白序列作比对。由于这些比对通常都希望能够推测新蛋白的功能,不管它们是双重比对还是多序列比对,都可以回答大量的其它的生物学问题。举例来说,面对一堆搜集的比对序列,人们会研究隐含于蛋白之中的系统发生的关系,以便于更好地理解蛋白的进化。人们并不只是着眼于某一个蛋白,而是研究一个家族中的相关蛋白,看看进化压力和生物秩序如何结合起来创造出新的具有虽然不同但是功能相关的蛋白。研究完多序列比对中的高度保守区域,我们可以对蛋白质的整个结构进行预测,并且猜测这些保守区域对于维持三维结构的重要性。 显然,分析一群相关蛋白质时,很有必要了解比对的正确构成。发展用于多序列比对的程序是一个很有活力的研究领域,绝大多数方法都是基于渐进比对(progressive alignment)的概念。渐进比对的思想依赖于使用者用作比对的蛋白质序列之间确实存在的生物学上的或者更准确地说是系统发生学上的相互关联。不同算法从不同方面解决这一问题,但是当比对的序列大大地超过两个时(双重比对),对于计算的挑战就会很令人生畏。在实际操作中,算法会在计算速度和获得最佳比对之间寻求平衡,常常会接受足够相近的比对。不管最终使用的是什么方法,使用者都必须审视结果的比对,因为再次基础上作一些手工修改是十分必要的,尤其是对保守的区域。 由于本书偏重于方法而不是原理,这里只讨论一小部分现成的程序。我们从两个多序列比对的方法开始,接下去是一系列的利用蛋白质家族中已知的模体或是式样的方法,最后讨论两个具有赠送的方法,因为绝大多数公开的算法不能达到出版物的数量。在本章结尾部分将会列出更详细的多序列比对的算法。 渐进比对方法 CLUSTAL W CLUSTAL W算法是一个最广泛使用的多序列比对程序,在任何主要的计算机平台上都可以免费使用。这个程序基于渐进比对的思想,得到一系列序列的输入,对于每两个序列进行双重比对并且计算结果。基于这些比较,计算得到一个距离矩阵,反映了每对序列 Bioinformatics: A Practical Guide to the Analysis of genes and Proteins Edited by A.D. Baxevanis and B.E.E. Ouellette ISBN 0-471-191965. pages 172-188. Copyright ? 1998 Wiley – Liss. Inc.