神经氨酸酶抑制剂抗流感病毒的研究进展_陈宝龙_邓旭_曾光尧_郭虹_周应军

作者简介:陈宝龙,男,硕士研究生研究方向:天然产物的合成与结构修饰
*
通信作者:周应军,男,教授研究方向:天然药物化学、
天然产物的合成与结构修饰
Tel :133********
E-mail :fisher203@https://www.360docs.net/doc/3a1825479.html,
神经氨酸酶抑制剂抗流感病毒的研究进展
陈宝龙,邓旭,曾光尧,郭虹,
周应军*
(中南大学药学院,长沙410013)
摘要:
神经氨酸酶(NA )是流感病毒表面一种蘑菇云状四聚体结构的包膜糖蛋白,
其抑制剂对高致病性流感病毒的各亚型均具有抑制作用,且其安全性和耐药性良好,可用于流感病毒的预防和治疗。笔者在归纳总结近年来该领域国内外文献的基础之上,对神经氨酸酶及其抑制剂的分类、构效关系以及国内外研究现状进行总结,有助于我们更好地利用现有条件设计并合成出活性更好、选择性更高的抗流感药物。关键词:流感病毒;神经氨酸酶抑制剂;分类;构效关系doi :10.11669/cpj.2015.01.002
中图分类号:R965
文献标志码:A
文章编号:1001-2494(2015)01-0007-08
Advances in Anti-Influenza Virus of Neuraminidase Inhibitors
CHEN Bao-long ,DENG Xu ,ZENG Guang-yao ,GUO Hong ,ZHOU Ying-jun *(School of Pharmaceutical Science ,Cen-tral South University ,Changsha 410013,China )ABSTRACT
Neuraminidase (NA )is a mushroom-shaped and tetramer structural envelope glycoprotein on the surface of the in-
fluenza virus.NA inhibitors can inhibit highly pathogenic influenza virus subtypes and have good safety and drug resistance ,hence they are widely used for the prevention and treatment of influenza virus.Based on the present domestic and foreign literatures in this field ,this paper summarizes the research status of neuraminidase inhibitors classification and structure-activity relation-ships.It will help us make better use of existing conditions to design and synthesize better active and more selective anti-influenza drugs.
KEY WORDS :influenza virus ;neuraminidase inhibitor ;classification ;structure-activity relationship
流感是由病毒引起的一种急性呼吸道传染性疾病,据相关统计全球每年约有25万 50万人死于流感及其并发症。近年来,随着全球物种活动范围的加大,流感病毒变异性的增强以及高致病性禽流感在世界范围内的频繁暴发,给人类的日常生活和经济发展带来了严重影响。目前,疫苗接种和药物治疗是防治流感的主要措施。但流感病毒亚型多、易突变的特点使得人们预测流感爆发的准确性大为降低,从而导致常规流感疫苗对不可预见的新型流感病毒束手无策。因此,保护人类健康的第一道防线、有效对抗流感病毒的只能是高效的抗流感药物。目前已上市的抗流感病毒药物包括2种类型:一类是M2蛋白抑制剂,包括金刚烷胺(1)和金刚乙胺(2)(图1),但这类药物仅对甲型流感病毒有效,而且耐药株的致病性和传染性以及严重的中枢神经系统副作用等均限制了此类药物的应用
[1]
。另一类是神经氨酸酶(neura-
minidase )抑制剂,包括扎那米韦(3)、奥司他韦(4)、帕拉米韦(5)、拉尼米韦(6)(图1)等,这类药物对高致病性流感病毒的各亚型均具有抑制作用,且其安全性和耐药性良好,可用于流感病毒的预防和治疗。笔者对神经氨酸酶及其抑制剂的分类、构效关系以及国内外研究现状进行综述,以期促进新型抗流感药物的研发。
1神经氨酸酶及其抑制剂靶点概述1.1
神经氨酸酶
神经氨酸酶是流感病毒表面一种蘑菇云状四聚体结
构的包膜糖蛋白,其活性中心位于各个亚基中央较深的口袋内。神经氨酸酶与流感病毒的复制和传播过程关系密切:首先,神经氨酸酶能够通过水解唾液酸与细胞之间的糖苷键来促进病毒在上呼吸道的传播和新一代病毒的释放。其次,神经氨酸酶可以将子代病毒表面的唾液酸残基清除,从而防止子代病毒因血凝素与唾液酸之间的相互作用而发生聚集。因此,神经氨酸酶抑制剂可以通过阻断病毒的生命周期,有效控制病毒在呼吸道的进一步传播[2]
。
1.2
神经氨酸酶活性中心
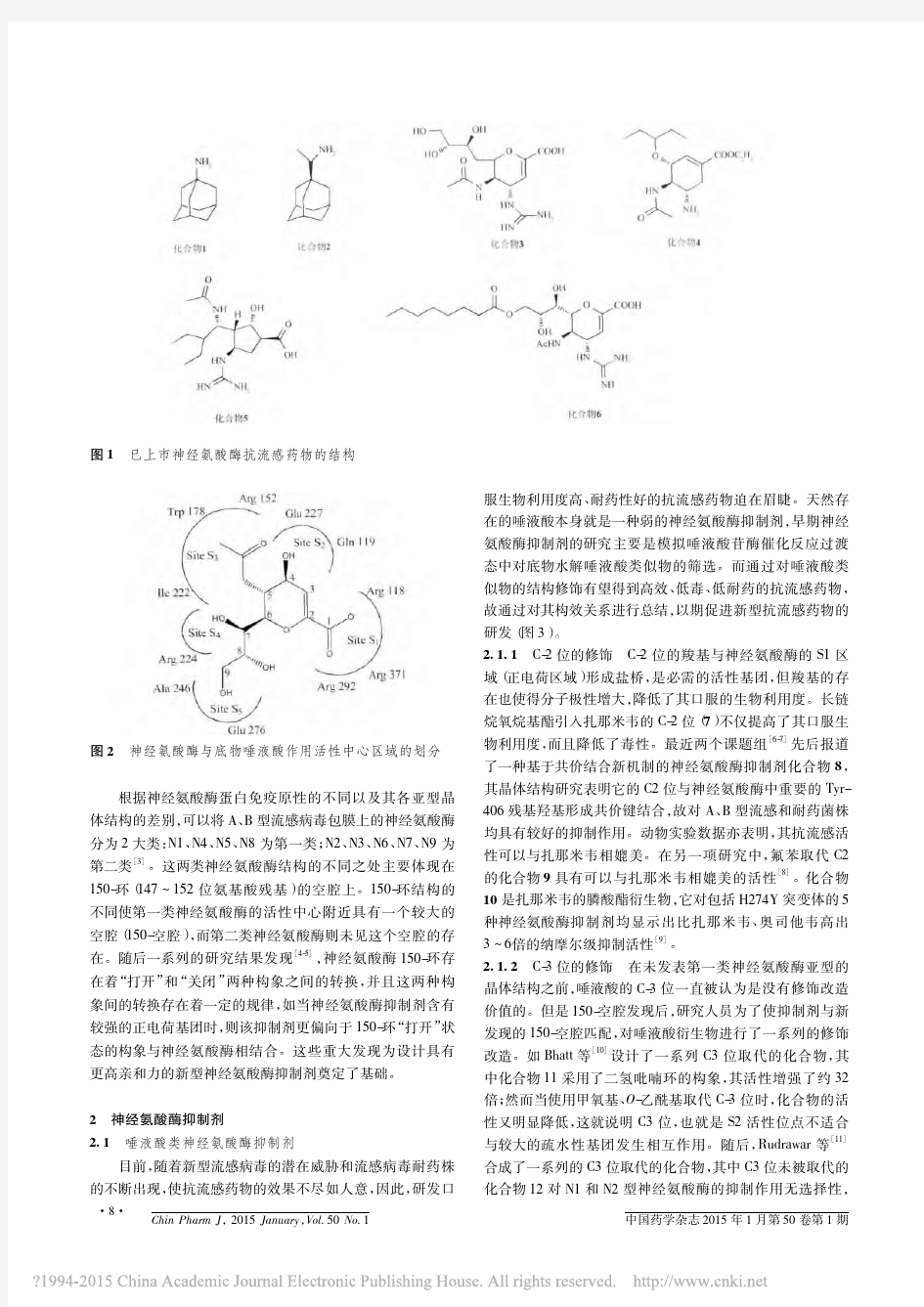
神经氨酸酶活性中心的框架是由18个保守的氨基酸残
基构成,其中8个高度保守的氨基酸残基可直接与底物水解唾液酸发生相互作用,影响整个水解糖苷键的催化过程,而其余的氨基酸残基则具有维持酶活性中心空间构象的作用。2003年,Stoll 建立了神经氨酸酶活性中心与其抑制剂的结合模型(图2),根据此模型,神经氨酸酶的活性中心可以分为5个结合区域(S1 S5)。
图1
已上市神经氨酸酶抗流感药物的结构
图2神经氨酸酶与底物唾液酸作用活性中心区域的划分根据神经氨酸酶蛋白免疫原性的不同以及其各亚型晶
体结构的差别,可以将A 、B 型流感病毒包膜上的神经氨酸酶分为2大类:N1、N4、N5、N8为第一类;N2、N3、N6、N7、N9为第二类
[3]
。这两类神经氨酸酶结构的不同之处主要体现在
150-环(147 152位氨基酸残基)的空腔上。150-环结构的不同使第一类神经氨酸酶的活性中心附近具有一个较大的空腔(150-空腔),而第二类神经氨酸酶则未见这个空腔的存在。随后一系列的研究结果发现
[4-5]
,神经氨酸酶150-
环存在着“打开”和“关闭”两种构象之间的转换,并且这两种构象间的转换存在着一定的规律,如当神经氨酸酶抑制剂含有较强的正电荷基团时,则该抑制剂更偏向于150-环“打开”状态的构象与神经氨酸酶相结合。这些重大发现为设计具有更高亲和力的新型神经氨酸酶抑制剂奠定了基础。2神经氨酸酶抑制剂
2.1
唾液酸类神经氨酸酶抑制剂
目前,随着新型流感病毒的潜在威胁和流感病毒耐药株的不断出现,使抗流感药物的效果不尽如人意,因此,研发口
服生物利用度高、耐药性好的抗流感药物迫在眉睫。天然存在的唾液酸本身就是一种弱的神经氨酸酶抑制剂,早期神经氨酸酶抑制剂的研究主要是模拟唾液酸苷酶催化反应过渡态中对底物水解唾液酸类似物的筛选。而通过对唾液酸类似物的结构修饰有望得到高效、低毒、低耐药的抗流感药物,故通过对其构效关系进行总结,以期促进新型抗流感药物的研发(图3)。2.1.1
C-2位的修饰
C-2位的羧基与神经氨酸酶的S1区
域(正电荷区域)形成盐桥,是必需的活性基团,但羧基的存在也使得分子极性增大,降低了其口服的生物利用度。长链烷氧烷基酯引入扎那米韦的C-
2位(7)不仅提高了其口服生物利用度,而且降低了毒性。最近两个课题组
[6-7]
先后报道
了一种基于共价结合新机制的神经氨酸酶抑制剂化合物8,其晶体结构研究表明它的C2位与神经氨酸酶中重要的Tyr-406残基羟基形成共价键结合,故对A 、B 型流感和耐药菌株均具有较好的抑制作用。动物实验数据亦表明,其抗流感活性可以与扎那米韦相媲美。在另一项研究中,氟苯取代C2的化合物9具有可以与扎那米韦相媲美的活性
[8]
。化合物
10是扎那米韦的膦酸酯衍生物,它对包括H274Y 突变体的5种神经氨酸酶抑制剂均显示出比扎那米韦、奥司他韦高出3 6倍的纳摩尔级抑制活性[9]。2.1.2
C-3位的修饰
在未发表第一类神经氨酸酶亚型的
晶体结构之前,唾液酸的C-3位一直被认为是没有修饰改造价值的。但是150-空腔发现后,研究人员为了使抑制剂与新发现的150-空腔匹配,对唾液酸衍生物进行了一系列的修饰改造。如Bhatt 等
[10]
设计了一系列C3位取代的化合物,
其中化合物11采用了二氢吡喃环的构象,其活性增强了约32倍;然而当使用甲氧基、O -乙酰基取代C-3位时,化合物的活性又明显降低,这就说明C3位,也就是S2活性位点不适合与较大的疏水性基团发生相互作用。随后,
Rudrawar 等[11]
合成了一系列的C3位取代的化合物,
其中C3位未被取代的化合物12对N1和N2型神经氨酸酶的抑制作用无选择性,
但甲苯基取代的化合物13对N1亚型的抑制作用比N2亚型增强了约200倍,且它对2009年H1N1型的突变体H274Y 和Q136K同样有效。这些研究结果表明,对唾液酸类似物的
C-3位进行修饰,有望得到高选择性高强度的治疗H5N1流感的神经氨酸酶抑制剂。
2.1.3C-4位的修饰唾液酸C4位和C6位分别被氨基和氨基羰基取代的化合物14抗A型流感病毒的活性高于扎那米韦,这就说明C4位的胍基并不一定是活性所必需基团。如Ye等[12]报道的化合物15,其C4位是由天冬酰胺基取代,它不仅显示出对H5N1感染的MDCK细胞良好的抑制活性(IC
50
=2.72mmol·L-1),而且显著增强了对N2亚型流感的抑制,这可能和天冬酰胺基与N2亚型神经氨酸酶可以更好的结合有关。随后,Wen等[13]在C4位的胍基上进行拓展修饰,发现哌嗪基取代的化合物16显示出了很强的神经氨酸酶抑制活性以及针对H1N1型流感亚微摩尔级的抗流感活性。
2.1.4C-5位的修饰研究人员发现唾液酸的C-5位必须保持一定的立体构型才具有抗流感活性,而且可以选择性增强抑制B型流感病毒的活性。2012年,Suzuki等[14]增加唾液酸的C-5位链长,得到化合物17、18,它们都表现出了不错的神经氨酸酶抑制活性。因此,乙酰氨基同样具有改造修饰的空间。
2.1.5C-6位侧链Yamashita等[15]将扎那米韦分子中C-7位羟基醚化、C-9位羟基长烷基链酯化,分别得到化合物19(R-125489)、20(CS-8958),它们对A、B型流感病毒以及耐奥司他韦的流感病毒株均具有较强的抑制作用,这就是最近在日本上市的新型抗流感病毒抑制剂辛酸拉尼米韦的前身。辛酸拉尼米韦与其他抗流感药物相比肺部滞留时间更长,单次吸入即可获得显著疗效的特点,每周吸入1次即可有效预防流感[16]。研究人员通过对感染H5N1合并高细胞因子血症的患者治疗观察发现,C7位羟基取代的扎那米韦具有纳摩尔级的神经氨酸酶抑制活性以及良好的抗炎活性[17]。因此,Liu等将咖啡酰基引入扎那米韦的C7位得到化合物21、22,他们均表现出了良好的抗流感和抑制炎症细胞因子的活性。Weight等[18]在扎那米韦的C7位进行修饰发现,化合物24的结构中多引入了1个L-谷酰胺聚合物的结构,这使得其抗流感活性比化合物23增强了约15 860倍。这些研究结果表明,唾液酸C6位侧链具有很大的结构改造空间。
2.2苯甲酸类神经氨酸酶抑制剂
与唾液酸结构和官能团类似的苯甲酸类衍生物因具有较少的手性中心、原料易为获取、合成方便等特点引起了科研工作者的关注(图4)。2008年,Zhang等[19]以对氨基水杨酸为原料,合成了一系列2位引入亲脂性基团的对氨基水杨酸衍生物,其中化合物26的活性最好,对H3N2的抑制活性
(IC
50
=32nmol·L-1)。随后,Yang等[20]运用柔性分子对接技术和分子动力学方法将化合物25与神经氨酸酶进行对接,结果发现化合物25与神经氨酸酶的活性中心吻合并形成了稳定的复合体系,更重要的是它与神经氨酸酶活性位点中保守的关键氨基酸残基之间存在着较强的静电和氢键作用,而与活性位点中易突变的氨基酸残基作用较弱,这为研制新型抗流感药物提供了很好的思路,同时也为修饰和改进苯甲酸类抑制剂提供了理论依据。2011年,Li等[21]基于天然产物异戊二烯基苯基醚母核合成了一系列的苯甲酸衍生物,结果发现化合物27对H1N1型流感的抑制活性比现有的苯甲酸类似物约高出了一个数量级,这是苯甲酸类神经氨酸酶抑制剂中前所未有的突破。2012年,Lalitha等[22]为了使苯环倾斜以获得与神经氨酸酶活性中心更好的结合,从而设计了化合物28,遗憾的是它并没有获得预期的神经氨酸酶抑制活性,这可能是由于苯环的倾斜导致其疏水性侧链与神经氨酸酶结合能力下降所导致的。
目前,虽然致力于苯甲酸类神经氨酸酶抑制剂研究的科研人员得到了一些活性较好的化合物,但是这一类抑制剂的研究始终没有获得用于临床的药物,这可能是由于苯环的平面刚性结构,使得其不易改变构象来适合神经氨酸酶的活性区域造成的。但是,值得我们注意的是该类化合物既非手性分子,又不含有强碱性基团,却仍然具有一定的神经氨酸酶抑制活性,因此,对这类化合物的改造可能发现不同作用机制的新型神经氨酸酶抑制剂。
2.3环己烯类神经氨酸酶抑制剂
相关研究发现,唾液酸母核环中的氧原子并未与神经氨酸酶的活性位点有任何作用力,因此研究人员将母核中的氧原子用碳原子替代得到了化合物29、30(图5)。这两个化合物结构上虽然只有双键的位置不同,但化合物29的活性比化合物30高出约40倍,故由此引入了环己烯类神经氨酸酶抑制剂的骨架。随后,研究人员以化合物29为先导化合物设计合成了GS4104(31),这就是已上市多年,站在抗流感前线的重要药物奥司他韦的前身,也是迄今为止环己烯类衍生物设计最成功的例子。2005年,研究人员提出以磷酸酯基取代奥司他韦母环上的羧基,这样既保持了与神经氨酸酶识别所需的负电基团,又增加了可以与血凝素结合的位点,从而有望设计出一类具有双重作用的神经氨酸酶抑制剂。随后,Wong等[23]在C-1位引入磷酸盐和酯(32,33),结果发现它们对H1N1的抑制活性比GS4071还要强,可以作为下一代神经氨酸酶抑制剂的先导化合物进行开发。2009年,Char-lotte等[24]对GS-4071的6位氨基进行修饰(34-37),发现这些化合物可以通过抑制神经氨酸酶与病毒膜蛋白的结合而起到抗流感的作用。最值得注意的是,化合物34具有很强的活性,是最有希望进入体内/体外评价的候选药物之一。同年,D'Souza等[25]运用分子建模技术将扎那米韦衍生物与神经氨酸酶进行分子对接后发现化合物38具有很好的活性,是最有希望开发成下一代神经氨酸酶抑制剂的候选物之一。2010年,Mohan等[26]设计合成出了一系列C-4位修饰的化合物,其中含三氮唑环的化合物39和含胍基的化合物40对神经氨酸酶均具有很好的抑制活性,活性可以和扎那米韦相媲美,可以作为潜在的神经氨酸酶抑制剂进行开发。2012年,Hussain[27]等人将乙烯酰氨基代替C-5位的乙酰氨
图3具有神经氨酸酶抑制活性的唾液酸类化合物类似物结构
图4具有神经氨酸酶抑制活性的苯甲酸类化合物类似物结构
图5具有神经氨酸酶抑制活性的环己烯类化合物类似物结构
基,使得化合物41比奥司他韦羧酸盐的活性增强了约2倍。Kongkamnerd等[28]在扎那米韦的结构基础上,对其C-5位进行修饰,发现化合物42和化合物43分别对N1和N3亚型神经氨酸酶的抑制活性均比扎那米韦强。
2.4环戊烷类神经氨酸酶抑制剂
研究人员发现,无论是扎那米韦的二氢吡喃环还是奥司他韦的环己烯环,在与神经氨酸酶结合的蛋白质晶体结构中都没有起到实质性的作用,仅仅是为神经氨酸酶抑制剂提供了一个类似于催化反应中近似平面结构过渡态的支架,反而环上的取代基显得更为重要。因此,研究人员将这些取代基移入其他类似环的结构,如环戊烷和吡咯烷环,以改善其抗流感活性以及口服生物利用度,这为新型的神经氨酸酶抑制剂设计奠定了基础(图6)。
研究人员将活性基团引入环戊烷骨架后发现化合物44、45对A、B型流感的抑制作用均强于扎那米韦和奥司他韦。但由于化合物45(BCX-1812,帕拉米韦)原料易得,成本较小,故对其进行了一系列的研究。临床研究表明,帕拉米韦能够有效抑制A、B型流感病毒,且对扎那米韦,奥司他韦耐药株同样有效,因此,2009年FDA根据美国疾病控制和预防中心的要求,已针对正在研究中的抗病毒药peramivir发布了一项紧急使用授权(EUA),先后在美国、日本上市。Bromba 等[29]为了研究帕拉米韦中胍基的作用,将帕拉米韦脱去胍基得到化合物46,虽然其活性略低于帕拉米韦,但是胍基的脱去降低了整个分子的极性,提高其生物利用度,这也许会成为比帕拉米韦效果更好的神经氨酸酶抑制剂。Brant等[30]报道了一种刚性双环砜结构的化合物47,其酶动力学研究表明,它对使用NP40灭活的A/Brisbane/59/2007(H1N1)病毒的抑制活性在4.5 700mmol·L-1之间,分子建模结果也表明其能够很好地覆盖羧基、胍基、乙酰氨基的结构功能。2.5吡咯烷类神经氨酸酶抑制剂
糖苷酶抑制剂中有一类化合物为亚氨基糖的结构,而神经氨酸酶抑制剂的骨架环只是催化反应近似平面结构中过渡态的支架,氨基的电正性又可以代替催化中的正离子,所以在吡咯烷类骨架化合物中可能发现强有效的神经氨酸酶抑制剂(图7)。
带有新颖的亲脂性基团的吡咯烷衍生物化合物48,对A、B型流感病毒神经氨酸酶均具有很好的抑制活性,从而成为了这类化合物的突破性进展。2008年,Krueger等[31]以
化合物48作为先导化合物,对其亲脂性部分进行了结构修饰,结果发现化合物49(A-315675)对A 和B 型流感病毒神经氨酸酶均表现出优于扎那米韦和奥司他韦的抑制活性。化合物50(A-322278)是将它做成羧酸乙酯的结构,使其口服生物利用度高达81.1%。2009年,
Baz 等[32]
使用野生型
H1N1的A 型流感病毒和耐奥司他韦的H274Y 突变型病毒进行A-322278活性测定,结果发现,A-322278与奥司他韦相比能够显著降低小鼠的死亡率和肺癌病毒滴度。目前A-322278正在进行临床前研究,有望成为强有效的神经氨酸酶抑制剂。最近报道了2个新的五元杂环神经氨酸酶抑制剂,噻唑烷骨架化合物51和脯氨酸结构改造化合物52[33]
,它们
对A 型流感的神经氨酸酶分别具有亚微摩尔和低微摩尔的
抑制活性,可开发为选择性治疗A 型流感的药物。2.6
天然产物神经氨酸酶抑制剂
新型流感病毒的潜在威胁和耐药流感病毒株的不断出现,严重威胁着人类的生命健康
[34]
(图8)。从植物中发现天
然抗病毒活性化学物质是开发抗病毒药物的重要途径,这也使得越来越多的研究人员希望从天然产物中找到一种新颖的神经氨酸酶抑制剂来解决流感病毒株耐药性的问题。
Ryu 等[35]研究发现了一系列烷基黄酮类化合物,其中化合物53具有纳摩尔级的抗流感活性(IC 50=380nmol ·L -1),而且研究表明它在动力学上属于非竞争性抑制剂,故其作用机制可能与现有的抗流感药物不同,有望开发成对现有药物耐药病毒株强有效的神经氨酸酶抑制剂。研究表明,羟基未被取代的黄酮神经氨酸酶抑制活性较弱[36]
,因此对
化合物53的结构进行改造可能发现强有效的神经氨酸酶抑
制剂。Dao 等
[37]
从甘草中分离出一种化合物54,它与奥司
他韦联用时,可显著增强奥司他韦对H1N1(H274Y )型流感病毒的抑制活性。研究结果间接的表明它抗流感的作用机制与奥司他韦不同,因此它具有对抗耐奥司他韦流感病毒的
潜力。
Xie 等[38]对具有抗病毒活性的天然产物咖啡酸进行研究,并筛选出一个活性较好的咖啡酸衍生物化合物55,它抑制N1、
N2型流感病毒的IC 50值分别为7.2和8.5μmol ·L -1,很有希望成为新型神经氨酸酶抑制剂的先导化合物。2012年,Dao 等[39]从姜黄中分离出一系列对H1N1和H9N2型病毒株具有强抑制活性的化合物,其中化合物56的活性最好,能够有效对抗新型甲型H1N1(WT )以及耐奥司他韦的新型甲型H1N1(H274Y 突变体)流感病毒。我们课题组将含有咖啡酰基的天然抗流感药物与神经氨酸酶进行对接发现,咖啡酰基并不和神经氨酸酶活性位点结合,因此这也间接的表明咖啡酰基的抗病毒能力来源于与其他活性区域进行作用。另外,课题组前期从广藿香中分离出了一系列的苯乙醇苷类化合物,其中含咖啡酰基的苯乙醇并β-吡喃葡萄糖结构具有很好的抗流感活性,有望作为先导化合物进行抗流感病毒药物的开发。3
当前抗流感治疗的机遇与挑战
随着对神经氨酸酶晶体结构研究的不断深入,其活性位点也不断为人们所了解,设计并合成强效抑制神经氨酸酶化合物的工作较之以前亦越来越清晰,这为找到强有效的抗流感药物打下了坚实的基础。
但是我们开发新型抗流感药物
图6
具有神经氨酸酶抑制活性的环戊烷类化合物类似物结构
图7
具有神经氨酸酶抑制活性的吡咯烷类化合物类似物结构
图8具有神经氨酸酶抑制活性的天然产物结构
的形势依旧严峻,唯一可以口服给药的抗流感药物奥司他韦也有可能因病毒株耐药性的不断增长而退出历史舞台。新型流感和耐现有抗流感药物病毒株的不断出现,又使得新一次全球范围内大型流感的发生埋下了伏笔,因此过分的依赖于现有抗流感药物并不能满足我们的临床需求。如何使唾液酸类抑制剂与神经氨酸酶的活性位点强有力的结合已经研究的比较透彻,因此提高现有治疗药物口服给药的生物利用度、寻找可以针对治疗高致病性流感的药物以及挖掘抗流感新的靶点是当务之急。从笔者介绍的内容可以发现,非竞争性抑制剂和新型骨架的抗流感化合物的筛选和修饰,可能会给我们带来强有效高选择性的抗流感药物,例如本课题组从抗流感中药广藿香中分离的一种苯乙醇苷类化合物就具有很好的抗流感活性,我们发现很多天然抗流感成分均具有咖啡酰基的类似片段,故课题组正在对其进行结构修饰,为我们拥有新一代抗流感药物提供了很大的希望。
REFERENCES
[1]ZHANG J,WANG Q,XU W F,et al.Synthesis and preliminary activity study design,pyrrolidine derivatives as neuraminidase in-
hibitors[J].Chin Pharm J(中国药学杂志),2008,43(4):
314-318.
[2]ZHANGRT,JIANG S B,LIU S W,et al.Progress inhibitors taregeting the influenza virus hemaggwtinin enter[J].Chin
Pharm J(中国药学杂志),2010,45(16):1208-1212.
[3]WANG M,QI J,LIU Y,et al.Influenza A virus N5neuraminidase has an extended150-cavity[J].J Virol,2011,85(16):8431-
8435.
[4]WANG P,ZHANG J Z.Selective binding of antiinfluenza drugs and their analogues to‘open'and‘closed'conformations of H5N1
neuraminidase[J],J Phys Chem B,2010,114(40):12958-
12964.
[5]LANDON MR,AMARORE,BARONR,et al.Novel drug gable hot spots in avian influenza neuraminidase H5N1revealed by
computational solvent mapping of a reduced and representative re-
ceptor ensemble[J].Chem Biol Drug Des,2008,71(2):106-
116.
[6]KIM J H,RESENDER,WENNEKES T.Mechanism-based cova-lent neuraminidase inhibitors with broad-spectrum influenza anti-
viral activity[J].Science,2013,340(6128):71-75.
[7]VAVRICKA C J,LIU Y,KIYOTA H,et al.Influenza neuramin-idase operates via a nucleophilic mechanism and can be targeted
by covalent inhibitors[J].Nat Commun,2013,4(7):1491.[8]FENG E,SHIN W J,ZHU X,et al.Structure-based design and synthesis of C-1-and C-4-modified analogs of zanamivir as neura-
minidase inhibitors[J].J Med Chem,2013,56(3):671-684.[9]SHIE J J,FANG J M,LAI P T,et al.A practical synthesis of zanamivir phosphonate congeners with potent anti-influenza activi-
ty[J].J Am Chem Soc,2011,133(44):17959-17965.
[10]BHATT B,BOHMR,KERRY P S,et al.Exploring the interac-tions of unsaturated glucuronides with influenza virus sialidase
[J].J Med Chem,2012,55(20):8963-8968.
[11]RUDRAWARS,KERRY PS,RAMEIX-WELTI M A,et al.
Synthesis and evaluation of novel3-C-alkylated-Neu5Ac2en deriv-
atives as probes of influenza virus sialidase150-loop flexibility
[J].Org Biomol Chem,2012,10(43):8628-8639.
[12]YE D,SHIN W J,LI N,et al.Synthesis of C-4-modified zanami-vir analogs as neuraminidase inhibitors and their anti-AIV activi-
ties[J].J Med Chem,2012,54(3):764-770.
[13]WEN W H,WANG S Y,TSAI K C,et al.Analogs of zanamivir with modified C4-substituents as the inhibitors against the group-1
neuraminidases of influenza viruses[J].Bioorg Med Chem,2010,
18(11):4074-4084.
[14]SUZUKI K,KOYAMA T,YINGSAKMONGKON S,et al.Synthesis and biological evaluation of sialic acid derivatives containing a long
hydrophobic chain at the anomeric position and their C-5linked
polymers as potent influenza virus inhibitors[J].Bioorg Med
Chem,2012,20(1):446-454.
[15]YAMASHITA M,TOMOZAWA T,KAKUTA M,et al.CS-8958,a prodrug of the new neuraminidase inhibitorR-125489,shows long-
acting anti-influenza virus activity[J].Antimicrob Agents Chemoth-
er,2009,53(1):186-192.
[16]KISO M,KUBO S,OZAWA M,et al.Efficacy of the new neura-minidase inhibitor CS-8958against H5N1influenza viruses[J].
PLoS Pathog,2010,6(2):e1000786.
[17]LIU K C,FANG J M,JAN J T,et al.Enhanced anti-influenza agents conjugated with antiinflammatory activity[J].J Med
Chem,2012,55(19):8493-8501.
[18]WEIGHT A K,HALDARJ,LVAREZ D C,et al.Attaching zan-amivir to a polymer markedly enhances its activity against drug-re-
sistant strains of influenza a virus[J].J Pharm Sci,2010,100
(3):831-835.
[19]ZHANG J,WANG Q,FANG H,et al.Design,synthesis,inhibito-ry activity,and SARstudies of hydrophobic paminosalicyclic acid
derivatives as neuraminidase inhibitors[J].Bioorg Med Chem
Lett,2008,16(7):3839-3847.
[20]YANG Z W,ZU Y G,WU X M,et al.Theory-(pentyloxy3-)Mechanism of benzoic acid and3neuraminidase[J].Acta Chem
Sin(化学学报),2010,68(14):1370-1378.
[21]LI J,ZHANG D M,ZHU X,et al.Studies on synthesis and struc-ture-activity relationship(SAR)of derivatives of a new natural
product from marine fungi as inhibitors of influenza virus neura-
minidase[J].Mar Drugs,2011,9(10):1887-1901.
[22]LALITHA V,ERIC SJ,GUNDURAO K,et al.Crystal structure of
a new benzoic acid inhibitor of influenza neuraminidase bound
with a new tilt induced by overpacking subsite C6[J].BMC
Struct Biol,2012,12(1):7.
[23]WONG C H,FANG J M,SHIE J J,et al.Synthesis of oseltami-vir containing phosphonate congeners with anti-influenza activity:
WO,2009029888[P].2005-05-03.
[24]CHARLOTTE D S,MEENA K,MAMATA J,et al.Search for novel neuraminidase inhibitors:Design,synthesis and interaction
of oseltamivir derivatives with model membrane using docking,
NMRand DSC methods[J].Biochim Biophys Acta,2009,1788
(9):1740-1751.
[25]D'SOUZA C,KANYALKARM,JOSHI,M,et al.Search for no-vel neuraminidase inhibitors:Design,synthesis and interaction of
oseltamivir derivatives with model membrane using docking,NMR
and DSC methods[J].Biochim Biophys Acta,2009,1788(9):
1740-1751.
[26]MOHAN S,MCATAMNEY S,HASELHORST T,et al.Carbocycles related to oseltamivir as influenza virus group-1-specific neuramini-
dase inhibitors.Binding to N1enzymes in the context of virus-like
particles[J].J Med Chem,2010,53(20):7377-7391.
[27]HUSSAIN BASHA S,PRASADRN.In-silico screening of ple-conaril and its novel substituted derivatives with neuraminidase of
H1N1influenza strain[J].BMCRes Notes,2012,5(1):105.[28]KONGKAMNERD J,CAPPELLETTI L,PRANDI A,et al.Syn-thesis and in vitro study of novel neuraminidase inhibitors against
avian influenza virus[J].Bioorg Med Chem,2012,20(6):2152-
2157.
[29]BROMBA C M,MASON J W,BRANT M G,et al.The deguanidi-nylated derivative of peramivir remains a potent inhibitor of influ-
enza neuraminidase[J].Bioorg Med Chem Lett,2011,21(23):
7137-7141.
[30]BRANT M G,WULFF J E.A rigid bicyclic platform for the gen-eration of conformationally locked neuraminidase inhibitors[J].
Org Lett,2012,14(23):5876-5879.
[31]KRUEGERA C,XU Y,KATI W M,et al.Synthesis of potent pyrrolidine influenza neuraminidase inhibitors[J].Bioorg Med
Chem Lett,2008,18(5):1692-1695.
[32]BAZ M,ABED Y,NNHME B,et al.Activity of the oral neuramin-idase inhibitor A-322278against the oseltamivir resistant H274Y
(A/H1N1)influenza virus mutant in mice[J].Antimicrob Agents
Chemother,2009,53(2):791-793.
[33]LIU Y,JING F,XU Y,et al.Design,synthesis and biological activity of thiazolidine-4-carboxylic acid derivatives as novel influ-
enza neuraminidase inhibitors[J].Bioorg Med Chem,2011,19
(7):2342-2348.
[34]GRIENKE U,SCHMIDTKE M,GRAFENSTEIN S V,et al.Influ-
enza neuraminidase:A druggable target for natural products[J].
Nat ProdRep,2012,29(1):11-36.
[35]RYU Y B,CURTIS-LONG M J,LEE J W,et al.Structural characteristics of flavanones and flavones from cudrania tricuspida-
ta for neuraminidase inhibition[J].Bioorg Med Chem Lett,2009,
19(17):4912-4915.
[36]LIU A L,WANG H D,LEE S M,et al.Structure-activity rela-tionship of flavonoids as influenza virus neuraminidase inhibitors
and their in vitro anti-viral activities[J].Bioorg Med Chem,
2008,16(15):7141-7147.
[37]DAO T T,NGUYEN P H,LEE H S,et al.Chalcones as novel in-fluenza A(H1N1)neuraminidase inhibitors from Glycyrrhiza in-
flate[J].Bioorg Med Chem Lett,2011,21(1):294-298.[38]XIE Y C,HUANG B,YU K X,et al.Caffeic acid derivatives:A new type of influenza Neuraminidase inhibitors[J].Bioorg Med
Chem Lett,2013,23(1):3556-3560.
[39]DAO T T,NGUYEN P H,WON H K,et al.Curcuminoids from Curcuma longa and their inhibitory activities on influenza A
neuraminidases[J].Food Chem,2012,134(1):21-28.
(收稿日期:2014-04-21)
中国药学会中药资源专业委员会成立大会暨新常态下中药资源可持续发展论坛在广西药用植物园隆重举行
2014年12月20日,由中国药学会主办,中国中医科学院中药资源中心、广西药学会和广西药用植物园承办的中国药学会中药资源专业委员会成立大会暨新常态下中药资源可持续发展论坛在广西药用植物园隆重举行。国家中医药管理局中药资源普查试点工作领导小组副组长李大宁,中国药学会副理事长兼秘书长丁丽霞,国家中医药管理局科技司中药科技处处长孙丽英及广西区相关部门领导出席会议并分别致辞。来自全国30个省、自治区、直辖市的107名专家及广西药用植物园全体科技人员参加了会议。成立大会由中国药学会副理事长、中国中医科学院副院长黄璐琦主持,中国药学会副理事长兼秘书长丁丽霞宣布了专业委员会主任委员、副主任委员名单,并颁发了聘书。会上,副主任委员缪剑华研究员表态专业委员会将凝聚所有委员的力量提升中药资源队伍的号召力、凝聚力和影响力,解决国内外中药资源发展的难点、重点问题,为企业、为社会、为人类做出贡献,在新常态下促进中药资源的可持续发展。李大宁副组长高度肯定了成立中药资源专业委员会的重要意义及作用,并针对中药资源的发展提出建议:希望有关部门及委员会在中药资源学术平台、中药资源科技创新体系、现代中药资源动态监测体系、科研与企业联盟、建立一批基地、建设一批重点实验室及重点学科等六个方面能够建立长效机制,达到中药资源可持续发展新常态化。
同期召开的论坛,以“新常态下中药资源可持续发展”为主题,中国中医科学院黄璐琦研究员、成都中医药大学彭成教授等14位全国知名专家就论坛主题作了精彩的报告,尤其是就如何解决当前中药资源存在的问题、如何更好地促进中药资源的可持续利用充分交流了各自的经验和观点。
随后,南京农业大学中药材研究所所长郭巧生教授主持了《药用植物栽培学》教材修订启动会,编委会编委及中药资源专业委员会委员参与修订。
此次大会成效显著,不仅团结和凝聚了全国中药资源研究的人才队伍,搭建了中药资源学术交流的平台,更有助于促进中药资源学科发展,指导、引领中药行业发展中药资源的保护、开发和利用。
doi:10.11669/cpj.2015.01.003
[本刊讯]
神经氨酸酶抑制剂的研究进展
上海应用技术学院 研究生课程(论文类)试卷 2 014 / 2 015学年第二学期 课程名称:新药研发与申报 课程代码:NX0702016 论文题目:神经氨酸酶抑制剂的研究进展 学生姓名:王震 专业﹑学号:化工1班,146061114 学院:化学与环境工程学院 课程(论文)成绩: 课程(论文)评分依据(必填): 1.论文结构规范,检索的文献资料经认真的综合分析整理,选材精简得当,条理清晰,语言流畅, 版面整洁美观。得分为90-100分。 2.论文结构较规范,检索的文献资料经分析整理,材料组织得当,条理清晰,语言流畅。得分为 80-89分。 3.论文结构基本规范,内容有小问题,检索的文献资料经一般性分类整理,条理较清晰,得分为 70-79分。 4.论文结构基本规范,内容未经认真整理,一般性罗列所检索的文献资料。得分为60-69分。 5.达不到上述第4点要求的论文,得分为0-59分。 任课教师签字: 日期:年月日
神经氨酸酶抑制剂的研究进展 摘要:2009年高致病性的H1N1流感大爆发,再次向人们敲响了警钟:随着毒株变异性的加强,流感疫苗已无力完全遏制疫情的传播[1]。我们知道,流感病毒在感染和传播过程中,作为其四大活性位点之一(其他三个是血凝素、M2离子通道和部分RNA聚合酶)的神经氨酸酶(NA)起到了重要作用。因此,抗流感病毒神经氨酸酶抑制剂的设计与合成势在必行。本文综述了抗流感病毒神经氨酸酶抑制剂(NAIs)的研究进展。 关键词:神经氨酸酶;变异;抑制剂;合成
The development of neuraminidase inhibitors Abstract: The pandemic of influenza virus in 2009 to human beings sounded the alarm: the influenza vaccine was feeling powerless to suppress the transmission of epidemic with the strengthening of strain’s variability. As we know, in the process of influenza virus’ infection and propagation, the neuraminidase, one of four neuraminiric active site (another active site,ie,Hemagglutinin,M2 ion channels and RNA polymerase), played a important role. Therefore, the designing and synthesis of anti-influenza virus neuramnidase inhibitors are imperative. And this paper reviewed the development of influenza-resistant virus neuraminidase inhibitors. Keywords: neuraminidase; variation; inhibitors; synthesis
流感病毒神经氨酸酶抑制剂的合理设计与筛选
流感病毒神经氨酸酶抑制剂的合理设计与筛选 摘要 流行性感冒(流感)是由流感病毒引起的上呼吸道疾病,每年影响数百万人的健康,造成比较严重的经济和社会问题。但是到目前为止,人类对流感病毒一直缺乏安全有效的控制手段,这使得抗流感病毒药物研究成为当前药学研究的一个热点。随着病毒学研究的进展,对流感病毒复制和感染过程的机理研究取得了重大的突破,在此基础上提出了一些可作为抗流感药物研究的靶标,比如:血凝素、神经氨酸酶、基质蛋白MZ以及核酸内切酶等。本文以其中的一种靶标化合物即神经氨酸酶为研究对象,对其抑制剂做出合理的设计及筛选,为研究与合衬抗流感病毒的药物提供一个较为合理的方向。 关键词:流感;流感病毒;神经氨酸酶;定量构效关系 1、立项依据 1.1、流感的危害以及防治现状 流行性感冒简称流感,是由流感病毒引起的呼吸道传染病,具有传染性强、流行面广、发病率高等特点,在儿童、老人及高危人群中的死亡率很高。流感感染后的症状主要表现为高热、咳嗽、流涕、肌痛等,多数伴有严重的心、肾等多种脏器衰竭并能导致死亡。流感可以通过消化道、呼吸道、皮肤损伤和眼结膜等多种途径传播,人员和车辆往来是传播本病的重要因素。 有数据表明,每次流感爆发期会使全球人口的近10%感染致病。仅在20世纪,流感的大流行就有三次,每次均使25%~35%的人感染致病,死亡率超过2%。迄今为止,世界上已发生过五次流感的大流行和若干次小流行,造成数十亿人发病,数千万人死亡,严重影响了人们的生活和社会经济的发展。 而预防和治疗流感给人们造成了沉重的经济负担,并导致劳动力的下降和人力资源的紧张。然而面对己对人类健康、社会经济造成严重破坏的流行性感冒,人类却一直缺乏有效的手段。 1.2、有神经氨酸酶抑制剂预防与治疗流感的现状 NA抑制剂是目前探索抗流感化学治疗药物研究中取得的突破性进展。它可以有效地阻断流感病毒的复制过程。与其它类型的抗流感病毒药物相比,NA抑制剂具有更高的疗效及更好的安全性和耐受性,并对所有的流感病毒亚型均有效,也很少出现病毒的抗药性。目前上市的NA抑制剂有两种:葛兰素公司得到Relenza罗氏公司的Tamiful,此外,还有一些神经氨酸酶抑制剂类药物正在开发中,如BioCryst公司的BANA-113、BANA-206;Abbott公司的A-315675等。由此可见,由于神经氨酸酶抑制剂类药物所具有的独特机制及疗效,它们己成为世界各大医药公司竞相研究的热点。
免疫抑制剂
免疫抑制剂的用药护理 免疫抑制剂定义 是一类通过抑制细胞及体液免疫反应,而使组织损伤得以减轻的化学或生物物质。其具有免疫抑制作用,可抑制机体异常的免疫反应,目前广泛应用于器官移植抗排斥反应和自身免疫性疾病的治疗。 免疫抑制剂的分类 1、钙调素抑制剂类:环孢菌素CsA类、他克莫司(FK506) 2、抗代谢类:硫唑嘌呤、霉酚酸脂(MMF) 3、激素类:甲强龙、醋酸泼尼松 4、生物制剂:抗T细胞球蛋白(ATG)、抗淋巴细胞球蛋白(ALG) 免疫抑制剂用药原则 1、预防性用药:环孢素A、FK506、霉酚酸脂(MMF)等。 2、治疗/逆转急性排斥反应(救治用药):MP(甲基强的松龙)、ALG或ATG、霉酚酸脂(MMF)、FK506等。 3、诱导性用药(因急性肾小管坏死而出现延迟肾功能、高危病人、二次移植、环孢素肾毒性病人):ATG、ALG等。 4、二联:激素(醋酸泼尼松)+抗代谢类(骁悉) 三联:激素(醋酸泼尼松)+抗代谢类(骁悉)+环孢素A(新山地明) 激素(醋酸泼尼松)+抗代谢类(骁悉)+FK506(他克莫司) 常用免疫抑制剂 1、环孢素(CsA):新山地明(进口)田可、赛斯平(国产) 作用机理
属于钙神经蛋白抑制剂,可以选择性抑制免疫应答,通过破坏使T细胞活化的细胞因子的表达,阻断参与排斥反应的体液和细胞效应机制,防止排斥反应的发生。 药物的吸收和代谢 新山地明受进食和昼夜节律的影响较山地明小,所以服药时间不必将用餐考虑在内。 环孢素A依靠胆汁排泄,肝功能障碍,胆汁淤积症或严重胃肠功能障碍都会影响环保素A的吸收和代谢。只有极少部分药物经肾脏排出,且不能经透析去除,所以对于肾脏功能不全者和需透析治疗的患者,均不需调整药物浓度。 副作用 (1)肾毒性:血清肌酐、尿素氮增高;肾功能损害。个体差异大,临床表现不典型,与其他原因引起的移植肾损害很难鉴别。且发生肾损害时,血药浓度可能正常,甚至偏低。 (2)接近半数的患者会出现肝脏毒性,其发生率与用药量密切相关。 (3)神经毒性:表现为肢体震颤、失眠、烦躁等。 (4)胃肠道反应:恶心、呕吐。 (5)其他并发证:高血压、高胆固醇血症、高钾血症、牙龈增生、糖尿病、多毛症。 用量 联合用药时:初始剂量为6~8mg/kg/d,Q12h,以后根据血药浓度调整。 注意事项 (1)严格按医嘱服药,定时服药,禁忌自行调整用药剂量。
防控流感的药物治疗方案
防控流感的药物治疗方案 今冬流感来袭,来势汹汹,全民抗击。据全国流感监测报告,此次冬季流感活动强度要强于往年,主要由甲型、乙型流感病毒引起,曾誉为“万能药”的板蓝根在此次流感中也并未获得有效治疗效果。1月9日,国家卫计委针对今冬流感的诊断和治疗发布《流行性感冒诊疗方案(2018年版)》(下称方案),给出了权威的建议,10余种抗病毒药物、中成药和中药饮片等被列入方案。我院药学部根据国家相关要求,加强了流感防控工作的部署,细心梳理相关在院药品,针对地区流感趋势及医院收治病患特点,在口服奥司他韦紧缺的情况下,积极与配送公司联系,加班加点、积极备药并提供合理用药建议及应对方案。科学安排、精心组织、克服困难,全力做好药学服务,努力满足临床及患者需求,积极应战流感袭击。 根据卫计委公布的常用药目录,目前我院已储备抗流感药物有: 抗病毒药:奥司他韦胶囊、奥司他韦颗粒、帕拉米韦氯化钠注射液。 中成药:莲花轻瘟胶囊、清开灵胶囊、疏风解毒胶囊、银黄胶囊、小儿小儿豉翘清热颗粒。 在此次流感肆虐中,应充分弘扬中医药文化,利用中医辩证理论进行治疗。在中药治疗流感方面,除奥司他韦外,也可使用方案中所列的中成药:疏风解表、清热解毒类的金花清感颗粒、连花清瘟胶囊、清开灵颗粒(口服液)、疏风解毒胶囊,以及银翘解毒类、桑菊感冒类等。其中儿童可选要为儿童抗感颗粒,小儿豉翘清热颗粒等。 临床用药过程中可从上述药物中选择对此次流感流行株敏感的抗病毒药物,注意区分普通感冒和流行性感冒,及早、合理应用抗流感病毒药物,避免盲目或不恰当地一种药物,造成过度用药、供货紧张等情况。治疗过程中需根据患者病理生理情况综合考虑,对于儿童、老年人、妊娠妇女、重症患者应特别注意,加强监护。规范和加强流感的临床用药管理,保证治疗和预防流感的有效性,从而达到缓解流感症状、降低并发症的发生率,降低病死率的目的。针对具体临床合理用药相关问题,推荐如下: 一、抗病毒治疗 1.抗流感病毒治疗时机 发病48 h 内进行抗病毒治疗可减少流感并发症、降低住院患者的病死率、缩短住院时间,发病时间超过48 h 的重症患者依然能从抗病毒治疗中获益。 重症流感高危人群及重症患者,应尽早(发病 48h 内)给予抗流感病毒治疗,不必等待病毒检测结果;如果发病时间超过 48 h,症状无改善或呈恶化倾向时也应进行抗流感病毒治疗。
钙调磷酸酶抑制剂
钙调磷酸酶抑制剂 (一)器官移植排斥反应 1、移植排斥反应 人体的免疫系统对各种致病因子有着非常完善的防御机制,能够对细菌、病毒、异物、异体组织、人造材料等“异己成分”进行攻击、破坏、清除,这种复杂的免疫学反应是人体非常重要的一种保护机制。受者进行同种异体组织或器官移植后,外来的组织或器官等移植物作为一种“异己成分”被受者免疫系统识别,后者发起针对移植物的攻击、破坏和清除,这种免疫学反应就是移植排斥反应(transplant rejection)。移植排斥反应是影响移植物存活的主要因素之一。 移植排斥反应是非常复杂的免疫学现象,涉及细胞和抗体介导的多种免疫损伤机制,发生原因主要是受体和移植物的人类白细胞抗原HLA(human leucocyte antigen)不同。因此,供者与受者HLA的差异程度决定了排异反应的轻或重。除同卵双生外,二个个体具有完全相同的HLA 系统的组织配型几乎是不存在的,因此在供受者进行配型时,选择HLA配型尽可能地接近的供者,是减少异体组织、器官移植后移植排斥反应的关键 2、发病机制 排斥反应的发生机制主要包括细胞免疫和体液免疫两个方面。临床最常见的急性排斥反应主要由细胞免疫介导,而超急性排斥反应和慢性排斥反应主要由体液免疫介导。 (1)细胞介导的排斥反应 细胞免疫在急性排斥反应发生发展过程中起主导作用。移植物中供体的淋巴细胞和树突状细胞具有丰富的HLA-Ⅰ和Ⅱ类抗原,是诱发排斥反应的主要致敏原。在移植物植入受体后,随着移植物的血液循环重建,供者的HLA-Ⅰ和Ⅱ类抗原不可避免的暴露于受者的免疫系统,受者的免疫细胞识别外来抗原后,即可引发下述一系列免疫反应: CD8+细胞毒性T细胞前体细胞与供者HLA-Ⅰ类抗原结合后活化增殖为成熟的细 胞毒性T细胞,对移植物产生攻击效应;CD4+T辅助细胞识别供体HLA-Ⅱ类抗原,促使抗原递呈细胞释放IL-I,后者可促进T辅助细胞增殖和释放IL-2,IL-2可进一步促进T辅助细胞增殖并为细胞毒性T细胞的分化提供辅助信号;除了IL-2之外,TH
常见蛋白酶抑制剂
当前位置:生物帮 > 实验技巧 > 生物化学技术 > 正文 蛋白酶及蛋白酶抑制剂大全 日期:2012-06-13 来源:互联网 标签: 相关专题:解析蛋白酶活性测定聚焦蛋白酶研究新进展 摘要: 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度 恩必美生物新一轮2-5折生物试剂大促销! Ibidi细胞灌流培养系统-模拟血管血液流动状态下的细胞培养系统 广州赛诚生物基因表达调控专题 蛋白酶抑制剂 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度。由于蛋白酶抑制剂在液体中的溶解度极低,尤其应注意在缓冲液中加人蛋白酶抑制剂时应充分混匀以减少蛋白酶抑制剂的沉淀。在宝灵曼公司的目录上可查到更完整的蛋白酶和蛋白酶抑制剂表。 常用抑制剂 PMSF 1)抑制丝氨酸蛋白酶(如胰凝乳蛋白酶,胰蛋白酶,凝血酶)和巯基蛋白酶(如木瓜蛋白酶); 2)10mg/ml溶于异丙醇中; 3)在室温下可保存一年; 4)工作浓度:17~174ug/ml(0.1~1.0mmol/L); 5)在水液体溶液中不稳定,必须在每一分离和纯化步骤中加入新鲜的PMSF。 EDTA 1)抑制金属蛋白水解酶; 2)0.5mol/L水溶液,pH8~9;
钙蛋白酶抑制蛋白(Calpastatin)的研究进展
读书报告 钙蛋白酶抑制蛋白研究进展 汪超钟正泽杨飞云周晓蓉曹兰 (重庆市畜牧科学院重庆荣昌402460) 摘要:钙蛋白酶在宰后肉类成熟过程中通过降解肌肉蛋白质而提高肉嫩度,钙蛋白酶抑制蛋白是在细胞内广泛表达的、高效的、专一性抑制钙蛋白酶活性的蛋白质,因此引起了广大研究者的广泛关注。本文阐述了钙蛋白酶抑制蛋白的结构,生物学作用,营养等因素对钙蛋白酶抑制蛋白的影响及其活性测定方法。 关键词:钙蛋白酶抑制蛋白结构生物学作用活性测定 Research Progress of Calpastatin W ang chao, Zhong zhengzhe, Y ang feiyun, Zhou xiaorong ,Cao lan (Chongqing Academy of Animal Science,Rongchong 402460 ) Abstact: Calpain make a contribution to Meat Tenderization by degradation of protein in the postmortem meat tenderization process. Calpastatin , a special endogenous inhibitor expressed extensively in cell , has a special inhibition to calpain. Therefore ,This paper review the structure ,biologic function, influenced factors , separation and activity assay of calpastatin. Key words: Calpastatin. Structure , Biologic function, influenced factors , activity assay 钙蛋白酶(Calpain)是一种钙激活中性半胱氨酸内肽酶,分布于所有脊椎动物的肌细胞内部。钙蛋白酶家族包括μ-钙蛋白酶,m-钙蛋白酶和钙蛋白酶抑制蛋白(Calpastatin)等。钙蛋白酶在骨骼肌中通过涉及生肌细胞分化、激发肌原纤维蛋白周转来调控生长,同时在宰后肉类成熟过程中通过降解肌肉蛋白质而提高肉嫩度。钙蛋白酶被钙离子和钙蛋白酶抑制蛋白调控[1]。钙蛋白酶抑制蛋白是一种有着多种功能的内源抑制剂,它通过抑制钙蛋白酶而发挥作用。本文综述了钙蛋白酶抑制蛋白的结构、生物学作用、营养等因素对钙蛋白酶抑制蛋白的影响及其分离与活性检测方法等。 1 钙蛋白酶抑制蛋白的结构 肌肉组织中钙蛋白酶抑制蛋白的分子量为77 KDa,斯托克半径为6.8nm,含
常用的免疫抑制剂的使用以及注意事项#(精选.)
常用的免疫抑制剂的使用以及注意事项 2015-03-09 环孢素(CsA):新山地明、田可、赛斯平 作用机理 属于钙神经蛋白抑制剂,可以选择性抑制免疫应答,通过破坏使T 细胞活化的细胞因子的表达,阻断参与排斥反应的体液和细胞效应机制,防止排斥反应的发生。 目前,市面出售的环孢素A有两种:一种为进口的,为瑞士诺华制药(新山地明),其剂型主要是胶囊;另一种为国产的,其剂型有口服液和胶囊。国內已有华东医药(赛斯平)、华北制药(田可)等多家生产厂家。 药物的吸收和代谢 环孢素A依靠胆汁排泄,肝功能障碍,胆汁淤积症或严重胃肠功能障碍都会影响环保素A的吸收和代谢。只有极少部分药物经肾脏排出,且不能经透析去除,所以对于肾脏功能不全者和需透析治疗的患者,均不需调整药物浓度。 新山地明受进食和昼夜节律的影响较山地明小,所以服药时间不必将用餐考虑在内。 副作用 1、肾毒性:个体差异大,临床表现不典型,与其他原因引起的移植肾损害很难鉴别。且发生肾损害时,血药浓度可能正常,甚至偏低。
2、接近半数的患者会出现肝脏毒性,其发生率与用药量密切相关。 3、其他并发症的发生率高血压41%~82%,高胆固醇血症37%,高尿酸血症35%~52%,高钾血症55%,震颤12%~39%,牙龈增生7%~43%,糖尿病2%~13%,多毛症29%~44%。 用量 联合用药时:初始剂量为6~8mg/kg/日,分两次服用,以后根据血药浓度调整。 注意事项 1、严格按医嘱服药,禁忌自行调整用药剂量。 2、遵医嘱监测血药浓度。 3、药品储存在15~30度室温中,忌冷冻。 4、定时服药,养成良好的定时服药习惯。 5、环孢素有不同的剂型,不同的厂家,各药在同一患者体内的生物利用度不尽相同,故改换不同剂型、不同厂家的药物时一定要在医生的指导下进行,以免出现排斥反应或药物中毒。 6、接受本药治疗的母亲不应授乳。 7、过敏体质者慎用注射液。 8、因硝苯吡啶可引起齿龈增生,故在应用环孢素A期间,发生齿龈增生的患者应避免使用硝苯吡啶。 血药浓度 1、CsA谷值浓度(C0)
神经氨酸酶抑制剂抗流感病毒的研究进展_陈宝龙_邓旭_曾光尧_郭虹_周应军
作者简介:陈宝龙,男,硕士研究生研究方向:天然产物的合成与结构修饰 * 通信作者:周应军,男,教授研究方向:天然药物化学、 天然产物的合成与结构修饰 Tel :133******** E-mail :fisher203@https://www.360docs.net/doc/3a1825479.html, 神经氨酸酶抑制剂抗流感病毒的研究进展 陈宝龙,邓旭,曾光尧,郭虹, 周应军* (中南大学药学院,长沙410013) 摘要: 神经氨酸酶(NA )是流感病毒表面一种蘑菇云状四聚体结构的包膜糖蛋白, 其抑制剂对高致病性流感病毒的各亚型均具有抑制作用,且其安全性和耐药性良好,可用于流感病毒的预防和治疗。笔者在归纳总结近年来该领域国内外文献的基础之上,对神经氨酸酶及其抑制剂的分类、构效关系以及国内外研究现状进行总结,有助于我们更好地利用现有条件设计并合成出活性更好、选择性更高的抗流感药物。关键词:流感病毒;神经氨酸酶抑制剂;分类;构效关系doi :10.11669/cpj.2015.01.002 中图分类号:R965 文献标志码:A 文章编号:1001-2494(2015)01-0007-08 Advances in Anti-Influenza Virus of Neuraminidase Inhibitors CHEN Bao-long ,DENG Xu ,ZENG Guang-yao ,GUO Hong ,ZHOU Ying-jun *(School of Pharmaceutical Science ,Cen-tral South University ,Changsha 410013,China )ABSTRACT Neuraminidase (NA )is a mushroom-shaped and tetramer structural envelope glycoprotein on the surface of the in- fluenza virus.NA inhibitors can inhibit highly pathogenic influenza virus subtypes and have good safety and drug resistance ,hence they are widely used for the prevention and treatment of influenza virus.Based on the present domestic and foreign literatures in this field ,this paper summarizes the research status of neuraminidase inhibitors classification and structure-activity relation-ships.It will help us make better use of existing conditions to design and synthesize better active and more selective anti-influenza drugs. KEY WORDS :influenza virus ;neuraminidase inhibitor ;classification ;structure-activity relationship 流感是由病毒引起的一种急性呼吸道传染性疾病,据相关统计全球每年约有25万 50万人死于流感及其并发症。近年来,随着全球物种活动范围的加大,流感病毒变异性的增强以及高致病性禽流感在世界范围内的频繁暴发,给人类的日常生活和经济发展带来了严重影响。目前,疫苗接种和药物治疗是防治流感的主要措施。但流感病毒亚型多、易突变的特点使得人们预测流感爆发的准确性大为降低,从而导致常规流感疫苗对不可预见的新型流感病毒束手无策。因此,保护人类健康的第一道防线、有效对抗流感病毒的只能是高效的抗流感药物。目前已上市的抗流感病毒药物包括2种类型:一类是M2蛋白抑制剂,包括金刚烷胺(1)和金刚乙胺(2)(图1),但这类药物仅对甲型流感病毒有效,而且耐药株的致病性和传染性以及严重的中枢神经系统副作用等均限制了此类药物的应用 [1] 。另一类是神经氨酸酶(neura- minidase )抑制剂,包括扎那米韦(3)、奥司他韦(4)、帕拉米韦(5)、拉尼米韦(6)(图1)等,这类药物对高致病性流感病毒的各亚型均具有抑制作用,且其安全性和耐药性良好,可用于流感病毒的预防和治疗。笔者对神经氨酸酶及其抑制剂的分类、构效关系以及国内外研究现状进行综述,以期促进新型抗流感药物的研发。 1神经氨酸酶及其抑制剂靶点概述1.1 神经氨酸酶 神经氨酸酶是流感病毒表面一种蘑菇云状四聚体结 构的包膜糖蛋白,其活性中心位于各个亚基中央较深的口袋内。神经氨酸酶与流感病毒的复制和传播过程关系密切:首先,神经氨酸酶能够通过水解唾液酸与细胞之间的糖苷键来促进病毒在上呼吸道的传播和新一代病毒的释放。其次,神经氨酸酶可以将子代病毒表面的唾液酸残基清除,从而防止子代病毒因血凝素与唾液酸之间的相互作用而发生聚集。因此,神经氨酸酶抑制剂可以通过阻断病毒的生命周期,有效控制病毒在呼吸道的进一步传播[2] 。 1.2 神经氨酸酶活性中心 神经氨酸酶活性中心的框架是由18个保守的氨基酸残 基构成,其中8个高度保守的氨基酸残基可直接与底物水解唾液酸发生相互作用,影响整个水解糖苷键的催化过程,而其余的氨基酸残基则具有维持酶活性中心空间构象的作用。2003年,Stoll 建立了神经氨酸酶活性中心与其抑制剂的结合模型(图2),根据此模型,神经氨酸酶的活性中心可以分为5个结合区域(S1 S5)。
抗流感药物研究趋势
抗流感药物研究趋势 张志豪 流行性感冒简称流感,是人类还不能完全有效控制的世界性传染病,与疟疾、结核病并列为世界死亡人数最多的三种传染病。世界性流感首次大流行是在1889年-1890年,最先发现于俄国中亚的布哈拉(今乌兹别克),先传到彼得堡,再传到西欧,一年内席卷全球。德国某些城市发病率达40%-50%。 1957年甲2型流感大流行。当年2月流行于中国贵州西部,3月传播全国,4月从香港出境扩散于世界,共死亡几十万人。流行地区发病率约50%,病死率0.01%。 1968年甲3型流感大流行。国外认为7月发源于香港,7月-8月流行于中国大部分地区,其后播散于世界。发病率30%,病死率与1957年相近,仅法国就死了4万人。医疗条件最好的美国,1934年-1966年32年间流感死亡数亦达51.2万人,平均每年1.6万人。 流感病毒是一种RNA病毒,属于正粘病毒科(Orthomyxoviridae family)流感病毒属(Influenza virus)。根据流感病毒核蛋白(Nucleoprotein, NP)和基质蛋白(Matrix protein, MP)的抗原性不同可以将其分为A、B、C三型。流感病毒A型和B型属于流感病毒属,而流感病毒C型属于流感病毒C属。A型流感病毒能感染多种动物,包括人、禽、猪、马等,存在禽类的流感病毒又称为禽流感病毒(Avian Influenza Virus, AIV),因此所有的禽流感病毒都属于A型流感病毒;B型和C型则主要感染人,从猪中也曾分离到。 流感病毒根据其表面纤突血凝素(Hemagglutinin, HA)和神经氨酸酶(Neuraminidase, NA)抗原的不同,又可以分为不同亚型。 流感病毒中重要的结构蛋白有以下几种: 1.镶嵌在病毒囊膜表面的血凝素(HA)和神经氨酸酶(NA)。这两种糖蛋白与流感病毒结合宿主细胞,感染宿主细胞有关。 2.同样镶嵌于病毒囊膜上的少量的M2蛋白,这是一种PH依赖的质子通道,与宿主细胞中流感病毒遗传物质的释放息息相关。 3.位于囊膜之下,基质层中的M1蛋白。 4.核糖核蛋白符合体中,与负链RNA相结合的核蛋白NP和3种聚合酶PB2、PB1、PA。 这几种蛋白与流感病毒的复制,蛋白质的转录和翻译有着密切关系。 我们要设计有效的抗流感病毒药物,就要从这几类蛋白着手,了解其结构与功能,挖掘其潜在的抗病毒位点。 1865年巴斯德认识到他称之为“病毒”的微生物是传染病的病因。另一位德国细菌学家保罗·埃尔利希,杜撰了“魔术弹”这一短语用于描述他自己的伟大目标——发明特定药物来杀死引起特定疾病的细菌但不杀死患者。1910年埃尔利希发明了非那明,这是最初治疗梅毒的特效药,但副作用也十分可怕。 而后,1932年另一位德国化学家吉哈德·多玛克,发明了基于硫元素的化合物,它能杀灭引起血中毒的致命链球菌。在之后十年中,医生们能够从一大批新“磺胺”制剂中进行选择,足以对付很大范围的感染,从产褥热、肺炎直到淋病、脑膜炎。一次令人惊奇的偶然,盘尼西林出现了。1920年代,苏格兰细菌学家亚历山大·弗莱明发现葡萄球菌被培养皿上的一块霉菌所摧毁——这次偶然
常见蛋白酶抑制剂
蛋白酶及蛋白酶抑制剂大全 标签: 相关专题:解析蛋白酶活性测定聚焦蛋白酶研究新进展 摘要: 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度 恩必美生物新一轮2-5折生物试剂大促销! Ibidi细胞灌流培养系统-模拟血管血液流动状态下的细胞培养系统 广州赛诚生物基因表达调控专题 蛋白酶抑制剂 破碎细胞提取蛋白质的同时可释放出蛋白酶,这些蛋白酶需要迅速的被抑制以保持蛋白质不被降解。在蛋白质提取过程中,需要加入蛋白酶抑制剂以防止蛋白水解。以下列举了5种常用的蛋白酶抑制剂和他们各自的作用特点,因为各种蛋白酶对不同蛋白质的敏感性各不相同,因此需要调整各种蛋白酶的浓度。由于蛋白酶抑制剂在液体中的溶解度极低,尤其应注意在缓冲液中加人蛋白酶抑制剂时应充分混匀以减少蛋白酶抑制剂的沉淀。在宝灵曼公司的目录上可查到更完整的蛋白酶和蛋白酶抑制剂表。 常用抑制剂 PMSF 1)抑制丝氨酸蛋白酶(如胰凝乳蛋白酶,胰蛋白酶,凝血酶)和巯基蛋白酶(如木瓜蛋白酶); 2)10mg/ml溶于异丙醇中; 3)在室温下可保存一年; 4)工作浓度:17~174ug/ml(0.1~1.0mmol/L); 5)在水液体溶液中不稳定,必须在每一分离和纯化步骤中加入新鲜的PMSF。 EDTA 1)抑制金属蛋白水解酶; 2)0.5mol/L水溶液,pH8~9; 3)溶液在4℃稳定六个月以上;
4)工作浓度:0.5~1.5mmol/L. (0.2~0.5mg/ml); 5)加入NaOH调节溶液的pH值,否则EDTA不溶解。 胃蛋白酶抑制剂(pepst anti n) l)抑制酸性蛋白酶如胃蛋白酶,血管紧张肽原酶,组织蛋白酶D和凝乳酶; 2)1mg/ml溶于甲醇中; 3}储存液在4℃一周内稳定,-20℃稳定6个月; 4)1作浓度:0.7ug/ml(1umol/L) 5)在水中不溶解。 亮抑蛋白酶肽(leupeptin) 1)抑制丝氨酸和巯基蛋白酶,如木瓜蛋白酶,血浆酶和组织蛋白酶B; 2)lOmg/ml溶于水; 3)储存液4℃稳定一周,-20℃稳定6个月; 4)工作浓度0.5mg/ml。 胰蛋白酶抑制剂(aprotinin) 1)抑制丝氨酸蛋白酶,如血浆酶,血管舒缓素,胰蛋白酶和胰凝乳蛋白酶; 2)lOmg/ml溶于水,pH7~8 3}储存液4℃稳定一周,-20℃稳定6个月; 4)工作浓度:0.06~2.0ug/ml(0.01~0.3umol/L); 5)避免反复冻融: 6)在pH>12.8时失活。 蛋白酶抑制剂混合使用 35ug/ml PMSF…………………………………丝氨酸蛋白酶抑制剂 0.3mg/ml EDTA…………………………………金属蛋白酶抑制剂 0.7ug/ml胃蛋白酶抑制剂(Pepstatin)…………酸性蛋白酶抑制剂 0.5ug/ml亮抑蛋白肽酶(Leupeptin)……………广谱蛋白酶抑制剂
蛋白酶抑制剂选择指南
蛋白酶抑制剂选择指南 1 蛋白酶抑制剂选择指南 抑制剂 工作浓度 分子量 抑制蛋白酶种类 稳定性 AEBSF终浓度1mM MW:239.5不可逆的丝氨酸蛋白酶抑制剂,抑制胰蛋白酶,糜 蛋白酶,纤溶酶,凝血酶及激肽释放酶. 可溶于水,其pH7的水溶液在4o C可保持稳定1-2个月,在pH>8的情况下会发生缓慢水解 Aprotinins 抑肽酶终浓度2ug/ ml MW:6512 可逆的丝氨酸蛋白酶抑制剂,可抑制纤溶酶,激肽 释放酶,胰蛋白酶,糜蛋白酶,但不抑制凝血酶和 Factor Xa。 非常稳定,当pH>12.8时失去活性,可溶于 水(10mg/ml),-20o C下可长期保存 Bestatin终浓度10uM MW:308.4 可逆的丙氨酰-氨基肽酶抑制剂, 工作液可保存一天,1mM的甲醇贮存液在 -20o C可保存至少一个月 E-64 Protease Inhibitor终浓度10uM MW:357.4 不可逆的半胱氨酸酸蛋白酶抑制剂,抑制半胱氨酸 酸蛋白酶而不会影响其他酶的半胱氨酸残基,与小 分子量的巯基醇如beta-巯基乙醇不会产生反应, 具有高度特异性。工作液在正常pH值下可保持稳定数天,1mM的水溶液在-20o C可保存几个月 EDTA, 4Na终浓度10mM MW:380.2 金属蛋白酶的可逆性螯合物,可能同时影响其他金 属依赖性生物过程。其水溶液很稳定,其贮存液(pH8.5的0.5M 水溶液)在4o C可保存数月 Leupeptin, 半硫酸盐 亮抑酶肽(亮肽素) 终浓度100uM MW:493.6 可逆的丝氨酸及半胱氨酸蛋白酶制剂,可抑制胰蛋 白酶样蛋白酶及一些半胱氨酸蛋白酶如:Lys-C内 切蛋白酶,激肽释放酶,木瓜蛋白酶,凝血 酶,Cathepsin B及胰蛋白酶。 工作液的稳定期为数小时,贮存液(10mM 水溶液)在4o C时稳定期为一周,-20o C时 稳定期为一个月 Pepstatin A 终浓度1uM MW:685.9 可逆的天冬氨酸蛋白酶,可抑制胃蛋白 酶,Cathepain B&L,血管紧张肽原酶(renin)及以1mg/ml溶于甲醇,搅拌过夜可以 1mg/ml溶于乙醇,333mg/ml溶于6N的
抗流感药物靶点及其抑制剂
抗流感药物靶点及其抑制剂 流感病毒是一种负螺旋单链RNA病毒,属于正黏病毒科。根据病毒核蛋白(nucleoproteins,NP)及基质蛋白(matrix proteins,M1)的抗原决定簇不同,流感病毒被分为三类:甲型(A)、乙型(B)、丙型(C)。流感病毒颗粒结构大致相似(如图1),自内而外可分成核心、基质蛋白以及包膜三部分。病毒子通常呈圆形,长丝状。甲型和乙型流感病毒核酸有八个RNA节段,负责编码十种蛋白,包括血凝素(HA)、神经氨酸酶(NA)、酸性蛋白(PA)、碱性蛋白1(PB1)、PB2、核蛋白(NP)、基质蛋白(M1)、离子通道蛋白(M2)、非结构蛋白(NS1)、核输出蛋白(NEP或NS2)。此外,大多数甲型流感病毒还有线粒体靶向的寡聚PB1-F2蛋白[1],报道其与细胞凋亡以及病毒毒力有关。这些病毒RNA片段同NP 结合并缠绕形成病毒核糖核蛋白体(vRNP),vRNP再与三聚的RNA聚合酶(PA、PB1、PB2)结合形成核糖核苷酸,负责RNA的复制和转录,这种结合模式确保了病毒RNA对于核酸酶保持敏感。丙型流感病毒只有七个RNA节段。基因组分节段的特点为流感病毒高频率基因重配提供了条件。病毒核心被外部的脂蛋白膜包围,在脂膜上有基质蛋白M1,其是病毒颗粒的主要蛋白,并通过化学键结合到vRNP。M2蛋白为具有离子通道活性的跨膜蛋白。乙型流感病毒缺乏M2蛋白,但是一种叫做BM2的蛋白可以起到类似M2蛋白作用。病毒最外层的包膜是包裹基质蛋白的磷脂双分子层,该膜来源于宿主细胞的细胞膜。膜表面具有两类非常重要的“刺突”,即两种糖蛋白,HA和NA。乙型流感病毒表面抗原相对简单,仅有一种HA 和一种NA。对于甲型流感,根据病毒表面抗原HA及NA的不同,其可进一步细分为16个HA亚型(H1 ~ H16)、9个NA亚型(N1 ~ N9)[2]。 图1 甲型流感病毒结构模式图[3]
神经氨酸酶
神经氨酸酶 神经氨酸酶又称唾液酸酶是分布于流感病毒被膜上的一种糖蛋白,它具有抗原性,可以催化唾液酸水解,协助成熟流感病毒脱离宿主细胞感染新的细胞,在流感病毒的生活周期中扮演了重要的角色。在甲型流感病毒中,神经氨酸酶的抗原性会发生变异,这成为划分甲型流感病毒亚型的依据,在目前已知的甲型流感病毒中共有9种不同的神经氨酸酶抗原型。 [编辑本段] 结构 分布于流感病毒包膜表面的神经氨酸酶是一个四聚体,由四个结构完全相同的单体亚基组合而成,其中每两个亚基通过一个二硫键相互链接,每两对单体即四个单体组成一个四聚体。每一个单体由球形的头部和细长的颈部两部分组成,头部是神经氨酸酶的活性部位,颈部则负责将蛋白锚定在病毒包膜表面。四聚体蛋白通过纤细的颈部与包膜连接,形状犹如蘑菇。1983年人们通过X射线衍射实验测定了神经氨酸酶头部的三级结构。实验测定结构显示,神经氨酸酶的活性头部是由六个β片层围绕成的桶状结构,桶状结构的内部是该酶的催化中心。实验显示,在所有亚型甲型流感病毒和乙型流感病毒表面分布的神经氨酸酶之间,一级结构即氨基酸序列的同源性并不高,仅有30%的氨基酸残基是同源的,但是亚基催化中心的附近的一段10余个残基组成的序列却高度保守。 神经氨酸酶由病毒RNA第六节段编码,在每粒流感病毒表面分布大约100个。[编辑本段] 神经氨酸酶功能 神经氨酸酶四聚体飘带模型神经氨酸酶负责催化唾液酸与糖蛋白之间糖苷键的水解。流感病毒侵染宿主后其表面的血凝素与宿主上皮细胞表面的血凝素受体结合,进入细胞,其基因利用宿主细胞的资源进行复制和表达,最终重新组装成新的流感病毒颗粒,以出芽的形式突出宿主细胞,但是成熟的流感病毒与宿主细胞之间,仍然依靠血凝素分子末端的唾液酸残基与血凝素受体分子表面的糖基团以2-6或2-3糖苷键链接,这使得流感病毒无法立即脱离宿主细胞。神经氨酸酶负责催化水解这一重要的糖苷键,使成熟的病毒颗粒最终脱离宿主细胞,感染新的上皮细胞,造成流感病毒在患者体内的扩散。 [编辑本段] 抑制剂 神经氨酸酶是流感治疗药物的作用靶点之一,自从人类了解该酶的作用之后,便开始了针对该酶抑制剂的研究,目前已经有两种神经氨酸酶抑制剂上市,一种进入三期临床研究。
流感病毒药物作用机制最新进展
流感病毒作用机理及抗流感药物研究进展 S1130556 田玉伟 摘要:流感病毒是人类健康的一大威胁。应对流感病毒的主要方式是疫苗和药物治疗。对可能大规模爆发的流感疫情来讲, 药物治疗是最好的控制流感病毒传播的手段。本文主要从流感病毒致病机理及抗流感病毒药物研究最新进展方面进行阐述。 关键词:流感病毒;抗病毒作用机制;抗流感病毒药物; The mechanism of influenza virus and the development of anti-influenza virus agent Abstract : Influenza is a major threat to millions of people worldwide. Vaccines and antiviral agents are two main options available to reduce the impact of the influenza virus, while anti-influenza agents are the most effective means to prevent the transmission of the highly contagious virus and to treat the epidemics of disease. In this article, recent progress in the research of the action mechanisms and structure-activity relationships of these anti-influenza virus agents were reviewed. Keywords: influenza virus ;anti-viral mechanism;anti-influenza virus agent; 1 流感病毒生物学结构 流行性感冒病毒[1-3](influenza virus)简称流感病毒,属正粘病毒科(orthomyxoviridae),呈球状或丝状,是一种有包膜和分节段的单链、负链RNA病毒。它可分为甲、乙、丙3型。甲型流感病毒常以流行形式出现,特点是传染性强,发病率高,传播快,可引起爆发流行乃至世界大流行,并可在动物中引起流行和造成大量动物死亡。乙型流感病毒,常引起流感局部爆发,不引起大流行。丙型流感病毒主要以散在形式出现。 流感病毒的基因由8个单链RNA片段组成,分别为NA、HA、NP、M、NS、PB1、PB2和PA基因。它们编码10种蛋白:膜蛋白血凝素(Hemagglutinin HA),神经氨酸酶(Neuraminidase NA),基质蛋白(Matrix protein1 M1,核蛋白(Nucleoprotein NP),3种RNA依赖多聚糖(RNA-dependent RNApolymerase PB1、PB2和PA)离子通道蛋白(Ion channel protein M2)和非结构蛋白(Non-structural protein NS1和NS2)。
抗流感病毒药物神经氨酸酶抑制剂奥司他韦研究进展
@@[1] De Villiers EM. Human papillomavirus infection in skin cancers[J].Biomed Pharmacother, 1998, 52 ( 1 ): 26- 33. @@[2] Dorian KJ, Beatty E, Atterbury KE. Detection of her pes simplex virus by the Kodak surecell herpes Test [J]. J Clin Microbiol, 1990, 28 ( 9 ): 2117-2119. @@[3] Stanley M. HPV vaccines: are they the answer? [J].Br Med Bull, 2008, 88 ( 1 ): 59-74.@@[4]龚向东,叶顺章,张秀炎,等.1991~2001年我国性 病流行病学分析[J].中华皮肤科杂志,2002,35 (3): 178-182. @@[5]谭德友,李新,欧趣娴,等.164例尖锐湿疣患者 人乳头瘤病毒基因型的检测[J].中山大学学报(医 学科学版),2009, 30 (3S): 109-112. @@[6] H Wang, Y L Qiao. Human papillomavirus type-dis tribution in condylomata acuminata of mainland China: a meta-analysis[J]. International Journal ofSTD & AIDS, 2008, 19 ( 10 ): 680-684. @@[7] Munoz N, Bosch F X, Sanjose S, et al. Epidemio logic classification of human pap illomavirus types asso ciated with cervical cancer[J]. N Engl Med, 2003, 348 ( 6 ): 518-527. @@[8]吕蓉.尖锐湿疣患者免疫功能的研究进展[J].中国艾 滋病性病,2003,9 (3): 187-188. @@[9] Goncalves MA, Burattini MN, Donadi EA, et al. Risk factors associated with genital warts in HIV-positive Brazilian women[J].Tumori, 2003, 89 ( 1 ): 9-15.@@[10]沈红萍,吴仁根.CO2电灼联合卡介菌多糖核酸治 疗尖锐湿疣临床观察[J].当代医学,2008,14(5): 83-84. 2011-07-25 抗流感病毒药物神经氨酸酶抑制剂 奥司他韦研究进展 张玲魏绍静 [摘要]高致病性禽流感H 5N1病毒在亚州的爆发和2009新型H1N1病毒的全球性传播,提示设计与研发新型的抗流感药物尤为迫切。神经氨酸酶在病毒复制和传播中发挥重要作用,且活性中心高度保守,其抑制剂成为抗流感药物研发的热点。该文回顾了临床广泛使用的奥司他韦(oseltamivir)药动学、药效学及耐药性方面的研究进展,为此药今后的临床应用提出建议。 神经氨酸酶抑制剂;流感病毒;奥司他韦;研究进展 10.3760/cma.j.issn. 1007-1245.2011.22.002 广东省自然科学基金资助项目(8151006002000005) 516000 广州市第八人民医院研究所 万方数据