试验六双波长分光光度法

典型实验教学案例简介
案例x 双波长分光光度法测定复方磺胺甲噁唑片中两组分含量
药物的含量测定是药物质量控制的重要方面。紫外分光光度法以其准确度、灵敏度高,简便快速等特点,成为药物含量测定的重要方法。紫外法测定含量时,常选择被测组分的最大吸收波长最为测定波长,以提高检测的灵敏度。当用紫外法测定复方制剂多组分中一种组分的含量时,共存组分常常也会有吸收干扰,此时,消除干扰就成为准确测定的关键环节。双波长分光光度法作为计算分光光度法的一种,就是消除干扰、实现多组分含量同时测定的一种有效手段。本实验就以复方磺胺甲噁唑片为实验对象,通过双波长分光光度法测定其两组分的含量,从而达到学习双波长法的原理和操作的目的。
一、实验目的
1.掌握双波长分光光度法的测定原理。
2.熟悉双波长分光光度法在复方制剂分析中的应用。
二、实验原理
当吸收光谱重叠的a、b两组分共存时,若要消除a组分的干扰测定b组分,可在a组分的吸收光谱上选择两个吸收度相等的λ1和λ2,测定混合物的吸光度差值。然后根据ΔA值计算b的含量。
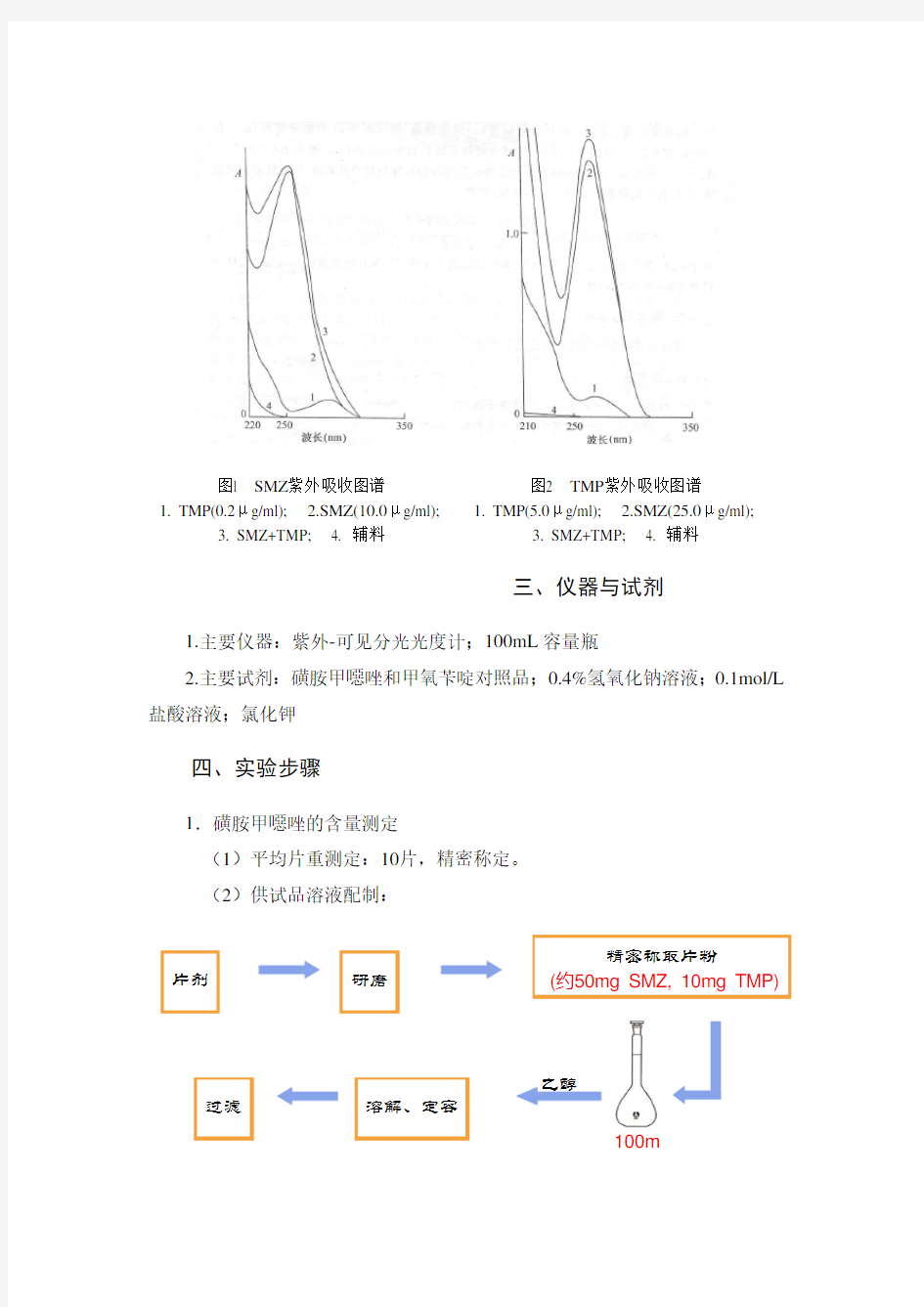
SMZ在257nm波长处有最大吸收,TMP在此波长吸收最小并在304nm波长附近有一等吸收点,故选定257nm为SMZ的测定波长(图1),在304nm波长附近选择参比波长。
TMP在239nm波长处有较大吸收,此波长又是SMZ的最小吸收峰,并在295nm波长附近有一等吸收点,故选定239nm为测定波长(图2),并规定在此波长附近选择供测定的参比波长。由于参比波长对测定影响较大,故采用对照品溶液来确定。此波长可因仪器不同而异,测定时应仔细选择。
三、仪器与试剂
1.主要仪器:紫外-可见分光光度计;100mL 容量瓶
2.主要试剂:磺胺甲噁唑和甲氧苄啶对照品;0.4%氢氧化钠溶液;0.1mol/L 盐酸溶液;氯化钾
四、实验步骤
1.磺胺甲噁唑的含量测定
(1)平均片重测定:10片,精密称定。 (2)供试品溶液配制:
图2 TMP 紫外吸收图谱
1. TMP(5.0μg/ml);
2.SMZ(25.0μg/ml);
3. SMZ+TMP;
4. 辅料
图l SMZ 紫外吸收图谱
1. TMP(0.2μg/ml);
2.SMZ(10.0μg/ml);
3. SMZ+TMP;
4. 辅料
精密称取片粉
(约50mg SMZ, 10mg TMP)
片剂 乙醇
溶解、定容
过滤 研磨
100m
(3)对照品溶液配制
(3)取对照品溶液(2)的稀释液,以257nm 为测定波长(λ2),在304nm 波长附近(每间隔0.5nm )选择等吸收点波长为参比波长(λ1),要求ΔA= A λ2
(2)
-A λ1(2)= 0,再在λ2与λ1两波长处分别测定供试品溶液的稀释液与对照品溶
液(1)的稀释液的吸收度,求出各自的吸收度差值(ΔA ),计算,即得。
2.甲氧苄啶的含量测定
(1)稀释液配制:取上述供试品溶液与对照品溶液(1)、(2)各5.00mL ,分别置于100mL 容量瓶中,各加HCl-KCl 溶液(0.1mol/LHCl 75mL + 6.9gKCl ,加水至1000mL ),稀释至刻度,摇匀。
(2)参比波长选定:取对照液(1)的稀释液,以239nm 为测定波长(λ2),在295nm 附近(每间隔0.2nm )选择等吸收点波长为参比波长(λ1) ,使ΔA=A λ2(1)-A λ1(1)= 0。
(3)双波长测定:在λ2与λ1两波长处分别测定供试品溶液的稀释液与对照品溶液(2)的稀释液的吸收度,求出各自的吸收度差值(ΔA ),计算,即得。
3.结果计算
100???????标示量
平均片重被测药物标示量%=W D Cs A A s
u
式中ΔA u 为供试液的ΔA 值,ΔA s 为对照液的ΔA 值,C s 为对照液浓度(mg/mL ),D 为稀释倍数,W 为取样量。
五、注意事项
乙醇
100mL
精称 105 ℃干至恒重
10mg TMP 对照品
50mg SMZ 对照品
乙醇
定容
定容 对照液(1) 对照液(2) 2.00ml
L
0.4%NaOH
100mL
2.00ml L
0.4%NaOH
1.测定之前应先检查仪器波长是否准确。
2.仔细寻找等吸收点波长
3.注意供试品溶液和对照品溶液的准确配制
六、思考题
1.双波长分光光度法是如何消除干扰的?
2.应用双波长分光光度法应如何选择测定波长和参比波长?
实验五--分光光度法测定甲醛
实验五:空气中甲醛的测定(酚试剂分光光度法) 实验目的: 掌握甲醛测定方法; 熟练掌握大气采样器和分光光度计的使用; 实验原理: 甲醛的测定方法:分光光度法、气相色谱法、酚试剂分光光度法、乙酰丙酮分光光度法; 空气中的甲醛与3-甲基2-苯并噻唑酮腙酚试剂反应生成嗪,嗪在酸性溶液中被高铁离子氧化形成蓝绿色化合物,颜色深浅与甲醛含量成正比,物质的最大吸收波长为630nm,通过比色定量。当采样体积为10L时最低检出质量浓度为0.01mg/m3。 实验仪器: 分光光度计(在630nm测定);大气采样器;具塞比色管(10ml);分析天平;滴定管;容量瓶;量筒;移液管等 1、吸收液原液:称量0.10g酚试剂[C6H4SN(CH3)C:NNH2·HCl,简称NBTH],加水溶解,倾于100ml具塞量筒中,加水到刻度。放冰箱中保存,可稳定三天。吸收液:量取吸收原液5ml,加95ml水,即为吸收液。采样时,临用现配。 2、1%硫酸铁铵溶液 3、碘溶液[C(1/2I2)=0.1000mol/L] 4、1mol/L氢氧化钠溶液 5、0.5mol/L硫酸溶液:取28ml浓硫酸缓慢加入水中,冷却后,稀释至1000ml。 6、硫代硫酸钠标准溶液[C(Na2S2O3)=0.1000mol/L] 0.5%淀粉溶液:将0.5g可溶性淀粉,用少量水调成糊状后,再加入100ml沸水,并煎沸2~3min至溶液透明确。 7、甲醛标准贮备溶液:取2.8ml含量为36~38%甲醛溶液,放入1L容量瓶中,加水稀释至刻度。此溶液1ml约相当于1mg甲醛。其准确浓度用下述碘量法标定。 实验步骤: 1、样品采集:用一个内装5ml吸收液的大型气泡吸收管,以0.5L/min流量,采气10L。并记录采样点的温度和大气压力。采样后样品在室温下应在24h内分析。 2、甲醛标准贮备溶液的标定:精确量取20.00ml待标定的甲醛标准贮备溶液,置于250ml 碘量瓶中。加入20.00ml[C(1/2I2)=0.1000mol/L]碘溶液和15ml 1mol/L氢氧化钠溶液,放置15min,加入0.5mol/L硫酸溶液,再放置15min,用[C(Na2S2O3)=0.1000mol/L]硫代硫酸钠溶液滴定,至溶液呈现淡黄色时,加入1ml 5%淀粉溶液继续滴定至恰使兰色褪去为止,记录所用硫代硫酸钠溶液体积(V2),ml。同时用水作试剂空白滴定,记录空白滴定所用硫化硫酸钠标准溶液的体积(V1),ml。甲醛溶液的浓度用公式(1)计算:甲醛溶液浓度(mg/ml)=(V1-V2)×N×15/20 (1) 式中:V1――试剂空白消耗[C(Na2S2O3)=0.1000mol/L]硫代硫酸钠溶液的体积,ml; V2――甲醛标准贮备溶液消耗[C(Na2S2O3)=0.1000mol/L]硫代硫酸钠溶液的体积,ml;N――硫代硫酸钠溶液的准确当量浓度; 15――甲醛的当量; 20――所取甲醛标准贮备溶液的体积,ml。 二次平行滴定,误差应小于0.05ml,否则重新标定。 绘制标准曲线: 用1.00μg/ml甲醛标准溶液,按下表制各标准色列管
实验分光光度法测定铁
实验分光光度法测定铁 The following text is amended on 12 November 2020.
实验十四邻二氮菲分光光度法测定铁的含量 一、实验目的 1.学习吸光光度法测量波长的选择方法; 2.掌握邻二氮菲分光光度法测定铁的原理及方法; 3. 掌握分光光度计的使用方法。 二、实验原理 分光光度法是根据物质对光选择性吸收而进行分析的方法,分光光度法用于定量分析的理论基础是朗伯比尔定律,其数学表达式为:A=εb C 邻二氮菲(又称邻菲罗啉)是测定微量铁的较好试剂,在pH=2~9的条件下,二价铁离子与试剂生成极稳定的橙红色配合物。摩尔吸光系数ε=11000 L·mol-1·cm-1。在显色前,用盐酸羟胺把Fe3+还原为Fe2+。 2Fe3++2NH 2OHHCl→2Fe2++N 2 +4H++2H 2 O+2Cl- Fe2+ + Phen = Fe2+ - Phen (橘红色) 用邻二氮菲测定时,有很多元素干扰测定,须预先进行掩蔽或分离,如钴、镍、铜、铅与试剂形成有色配合物;钨、铂、镉、汞与试剂生成沉淀,还有些金属离子如锡、铅、铋则在邻二氮菲铁配合物形成的pH范围内发生水解;因此当这些离子共存时,应注意消除它们的干扰作用。 三、仪器与试剂 1.醋酸钠:l mol·L-1; 2.盐酸:6 mol·L-1; 3.盐酸羟胺:10%(用时配制); 4.邻二氮菲(%):邻二氮菲溶解在100mL1:1乙醇溶液中; 5.铁标准溶液。 (1)100μg·mL-1铁标准溶液:准确称取(NH 4) 2 Fe(SO 4 ) 2 ·12H 2 0于烧杯中, 加入20 mL 6 mol·L-1盐酸及少量水,移至1L容量瓶中,以水稀释至刻度,摇匀. 6.仪器:7200型分光光度计及l cm比色皿。 四、实验步骤 1.系列标准溶液配制 (1)用移液管吸取10mL100μg·mL-1铁标准溶液于100mL容量瓶中,加入2mL 6 mol·L-1盐酸溶液, 以水稀释至刻度,摇匀. 此溶液Fe3+浓度为10μg·mL-1. (2) 标准曲线的绘制: 取50 mL比色管6个,用吸量管分别加入0 mL,2 mL,4 mL, 6 mL, 8 mL和10 mL10μg·mL-l铁标准溶液,各加l mL盐酸羟胺,摇匀; 经再加2mL邻二氮菲溶液, 5 mL醋酸钠溶液,摇匀, 以水稀释至刻度,摇匀后放置 10min。 2.吸收曲线的绘制 取上述标准溶液中的一个, 在分光光度计上,用l cm比色皿,以水为参比溶液,用不同的波长,从440~560 nm,每隔10 nm测定一次吸光度,在最大吸收波长
试验六双波长分光光度法
典型实验教学案例简介 案例x 双波长分光光度法测定复方磺胺甲噁唑片中两组分含量 药物的含量测定是药物质量控制的重要方面。紫外分光光度法以其准确度、灵敏度高,简便快速等特点,成为药物含量测定的重要方法。紫外法测定含量时,常选择被测组分的最大吸收波长最为测定波长,以提高检测的灵敏度。当用紫外法测定复方制剂多组分中一种组分的含量时,共存组分常常也会有吸收干扰,此时,消除干扰就成为准确测定的关键环节。双波长分光光度法作为计算分光光度法的一种,就是消除干扰、实现多组分含量同时测定的一种有效手段。本实验就以复方磺胺甲噁唑片为实验对象,通过双波长分光光度法测定其两组分的含量,从而达到学习双波长法的原理和操作的目的。 一、实验目的 1.掌握双波长分光光度法的测定原理。 2.熟悉双波长分光光度法在复方制剂分析中的应用。 二、实验原理 当吸收光谱重叠的a、b两组分共存时,若要消除a组分的干扰测定b组分,可在a组分的吸收光谱上选择两个吸收度相等的λ1和λ2,测定混合物的吸光度差值。然后根据ΔA值计算b的含量。 SMZ在257nm波长处有最大吸收,TMP在此波长吸收最小并在304nm波长附近有一等吸收点,故选定257nm为SMZ的测定波长(图1),在304nm波长附近选择参比波长。 TMP在239nm波长处有较大吸收,此波长又是SMZ的最小吸收峰,并在295nm波长附近有一等吸收点,故选定239nm为测定波长(图2),并规定在此波长附近选择供测定的参比波长。由于参比波长对测定影响较大,故采用对照品溶液来确定。此波长可因仪器不同而异,测定时应仔细选择。
三、仪器与试剂 1.主要仪器:紫外-可见分光光度计;100mL 容量瓶 2.主要试剂:磺胺甲噁唑和甲氧苄啶对照品;0.4%氢氧化钠溶液;0.1mol/L 盐酸溶液;氯化钾 四、实验步骤 1.磺胺甲噁唑的含量测定 (1)平均片重测定:10片,精密称定。 (2)供试品溶液配制: 图2 TMP 紫外吸收图谱 1. TMP(5.0μg/ml); 2.SMZ(25.0μg/ml); 3. SMZ+TMP; 4. 辅料 图l SMZ 紫外吸收图谱 1. TMP(0.2μg/ml); 2.SMZ(10.0μg/ml); 3. SMZ+TMP; 4. 辅料 精密称取片粉 (约50mg SMZ, 10mg TMP) 片剂 乙醇 溶解、定容 过滤 研磨 100m
栀子双波长融合高效液相色谱数字化指纹图谱研究
梔子双波长融合高效液相色谱数字化指 纹图谱研究 (作者: _________单位:____________ 邮编:___________ ) 【摘要】目的采用双波长融合法建立梔子254 nm和326 nm波长下的融合HPLC指纹图谱。方法采用反相高效液相色谱 法,使用CenturySIL C18 BDS 柱(20 cm >4.6mm , 5 町i),以1%醋酸水-1%醋酸乙腈为流动相,低压线性梯度洗脱,柱温(30.0 士0.15 )C,采用DAD检测器同时采集2个特征吸收波长下的信号,测定10批不同产地梔子HPLC指纹图谱。结果用“中药色谱指纹图谱超信息特征数字化评价系统”软件建立了梔子254 nm和326 nm的双波长融合HPLC指纹图谱,以梔子苷为参照物,确定了41个共有峰,并对其进行了全面评价。结论所建立的双波长融合谱能有效控制梔子的质量,克服了单波长检测时信息量有限的缺点,能较全面、真实地揭示中药材的内在质量特征。 【关键词】梔子;高效液相色谱数字化指纹图谱;双波长融合;超信息特征 Abstract : ObjectiveTo explore the HPLC-FPS of water extract
of Fructus Garde niae by dual-wavele ngth fusing method. MethodsThe chromatographic fin gerpri nts were obta ined by injecting the sample solution each time on a CenturySIL C18 BDS colu mn (20 cm X4.6 mm , 5 ^m) with the gradie nt eluti on solve nt system composed of 1% acetate acid -water and 1% acetate acid -acetonitrile. The flow rate was 1.0ml/min, the column temperature was maintained at (30.00 ±0.15) °C and the DAD detector was used. ResultsThe HPLC-FPS of water extract of Fructus Garde niae was established with dual-wavelength fusing method, acquired 41 com mon peaks by tak ing the peak of gen iposide as referen tial peak. The fin gerpri nts were comprehe nsively evaluated by The digitized evaluation system of the Traditional Chinese Medicine fingerprint with the super-information characteristics ” and the results were an alyzed and compared with that of the fin gerpri nts obta ined un der 265 nm. Con clusi on The dual-wavele ngth fusing method can effectively con trol the quality of Fructus Garde niae and overcome the low information of mono-wavelength detection. It offers guara ntee for the quality con trol of Fructus Garde niae and provides a new method for thoroughly and authentically revealing the characteristics of TCM. Key words : Fructus Gardeniae; Dual-wavelength fusing method; High performa nee liquid chromatography;F in gerpr ints;
紫外可见分光光度法测定水杨酸的含量[详实参考]
紫外可见分光光度法测定水杨酸的含量 一、实验目的 1、了解紫外可见分光光度计的性能、结构及其使用方法。 2、掌握紫外-可见分光光度法定性、定量分析的基本原理和实验技术。 二、实验原理 紫外-可见光谱是用紫外-可见光测获的物质电子光谱,它研究产生于价电子在电子能级间的跃迁,研究物质在紫外-可见光区的分子吸收光谱。当不同波长的单色光通过被分析的物质时能测得不同波长下的吸光度或透光率,以ABS为纵坐标对横坐标波长λ作图,可获得物质的吸收光谱曲线。一般紫外光区为190-400nm,可见光区为400-800nm。 紫外吸收光谱的定性分析为化合物的定性分析提供了信息依据。由于分子结构不同但只要具有相同的生色团,它们的最大吸收波长值就相同。因此,通过对末知化合物的扫描光谱、最大吸收波长值与已知化合物的标准光谱图在相同溶剂和测量条件下进行比较,就可获得基础鉴定。 利用紫外吸收光谱进行定量分析时,必须选择合适的测定波长。苯甲酸和水杨酸的紫外吸收光谱如图1所示。 图1 苯甲酸与水杨酸紫外吸收光谱图 1-苯甲酸;2-水杨酸 水杨酸在波长300 nm处有吸收峰,而苯甲酸此处无吸收,在波长230 nm两组吸收峰重叠,为了避开其干扰,选用300 nm波长作为测定水杨酸的工作波长。由于乙醇在250~350nm无吸收干扰,因此可用60%乙醇为参比溶液。 三、仪器与试剂 1.仪器 紫外-可见分光光度计(UVWIN 5,北京普析通用仪器有限公司);容量瓶
100mL 1个、50mL 5个;刻度吸量管1mL、2mL、5mL各1支。 2.试剂 水杨酸对照品(分析纯);60%乙醇溶液(自制)。 四、实验步骤 1、标准溶液的制备:准确称取0.0500 g水杨酸置于100 mL烧杯中,用60%乙醇溶解后,转移到100 mL容量瓶中,以60%乙醇稀释至刻度,摇匀。此溶液浓度为0.5mg·mL-1。 2、将五个50mL容量瓶按1-5依次编号。分别移取水杨酸标准溶液0.50、1.00、2.00、3.00、4.00mL于相应编号容量瓶中,各加入60%乙醇溶液,稀释至刻度,摇匀。 3、用1 cm石英吸收池、,以60%乙醇作为参比溶液,在200~350 nm波长范围内测定一份水杨酸标准溶液的紫外吸收光谱,确定最大吸收波长。 4、在选定波长下,以60%乙醇为参比溶液,由低浓度到高浓度测定水杨酸标准溶液系列及未知液的吸光度。以水杨酸标准溶液的吸光度为纵坐标,浓度为横坐标绘制标准曲线,根据水杨酸试液的吸光度,通过标准曲线计算水杨酸试样中水杨酸的含量。 表1 标准曲线制定及未知试样浓度检测
高效液相色谱—紫外双波长法同时测定玄参配方颗粒中3种主要成分的含量
高效液相色谱—紫外双波长法同时测定玄参配方颗粒中3种主要成 分的含量 目的建立高效液相色谱-紫外双波长法同时测定玄参配方颗粒中3种主要成分哈巴苷、哈巴俄苷和肉桂酸含量的方法。方法采用Ultimate AQ-C18色谱柱(250 mm×4.6 mm,5 μm),流动相为1%冰醋酸水溶液和乙腈,梯度洗脱,流速1.0 mL/min,检测波长前13 min为210 nm,13 min后为278 nm,柱温30 ℃。结果哈巴苷、哈巴俄苷、肉桂酸的线性范围分别为0.066 54~0.665 4 μg(r=1.000 0)、0.024 23~0.242 3 μg (r=0.999 9)、0.100 28~1.002 8 μg(r=0.999 9),平均加样回收率(n=6)分别为99.80%±1.22%、100.31%±1.30%、100.22%±1.24%。结论本法简单、灵敏度高、重复性好、准确可靠,可为玄参配方颗粒的质量控制以及标准研究提供参考依据。标签:玄参配方颗粒;哈巴苷;哈巴俄苷;肉桂酸;高效液相色谱-紫外双波长法 中药玄参为玄参科植物玄参Scrophularia ningpoensis Hemsl.的干燥根,始载于《神农本草经》,列为中品,为“浙八味”中药之一。玄参配方颗粒是在中医药理论指导下,采用现代生产工艺,以水作为提取溶媒,对单味玄参药材饮片进行煎煮、过滤、浓列检测器G1315D;ChemStation for LC 3D systems色谱工作站;Ultimate AQ-C18色谱柱(250 mm×4.6 mm,5 μm);Sartorius BP210S电子分析天平(德国);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司)。 哈巴俄苷对照品(批号111730-201106,含量以96.0%计,中国食品药品检定研究院),哈巴苷对照品(批号111729-201204,含量以94.5%计,中国食品药品检定研究院),肉桂酸对照品(批号110786-200503,中国药品生物制品检定所)。玄参配方颗粒(批号A1301311、A120847、A120696),培力(南宁)药业有限公司。乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。 2 方法与结果 2.1 色谱条件 色谱柱:Ultimate AQ-C18(250 mm×4.6 mm,5 μm);流动相:1%冰醋酸水溶液(A)-乙腈(B),梯度洗脱(0~10 min,5%~10%B;10~30 min,10%~30%B;30~35 min,30%~50%B;35~40 min,50%~60%B);流速:1.0 mL/min;柱温:30 ℃;检测波长:前13 min为210 nm,13 min后改为278 nm;进样量:10 μL。理论塔板数均≥5000,与相邻杂峰分离度均>1.5。色谱图见图1。
双波长法测定安钠咖中组分含量
院系:医学检验系 班级:11检验本科1班 姓名:**** 双波长法测定安钠咖中组分含量 实验目的和要求 1、掌握紫外可见分光光度计的基本操作; 2、掌握双波长分光光度法测定二元混合物中待测组分含量的原理和方法; 3、掌握在物质中吸收曲线上寻找吸收点、测定波长、参比波长的方法; 4、掌握标准曲线绘制及应用; 5、了解双波长分光光度法在单光束分光光度计上的测定方式。 实验原理 每毫升安钠咖注射液中含0.12g 无水咖啡因和0.13g 苯甲酸钠,要求二组分的含量均应为标示量的93%~107%。在0.10mol/L 盐酸溶液中,苯甲酸钠在230nm 波长处有一较强吸收峰,咖啡因在272nm 波长处有一较强吸收峰;二者吸收曲线重叠十分严重,直接采用吸光度进行定量测定时,相互之间有严重干扰。 根据光吸收定律,溶液吸光度应为各个组分吸光度的加合。当在230nm 波长处测定苯甲酸钠(此时把咖啡因视为干扰组分)时,测定的吸光度为: 咖啡因苯甲酸钠安钠咖230 230230A A A += 但是在咖啡因的吸收曲线可以发现,咖啡因在230nm 和257nm 两波长处得吸收相等(吸光度值相等,即等吸收点): 吸光度 波长 230nm
咖啡因咖啡因257 230A A = 因此,通过直接测定混合物溶液在230nm 和257nm 两波长处得吸光度值,再计算二吸光度的差值,可消除咖啡因对苯甲酸钠测定的干扰: )()(咖啡因苯甲酸钠咖啡因苯甲酸钠安钠咖安钠咖257257230230257230A A -A A A -A A ++==? 苯甲酸钠苯甲酸钠苯甲酸钠苯甲酸钠苯甲酸钠)(C b -b A -A 257 230257230εε== =KC 苯甲酸钠 可以看出,混合物溶液在230nm 和257nm 两波长处得吸光度差值仅与苯甲酸钠浓度成正比,而与咖啡因浓度无关,从而实现苯甲酸钠的定量分析。 同理,当在272nm 波长处测定咖啡因(视苯甲酸钠为干扰组分)时,可选择272nm 和253nm 两个苯甲酸钠的等吸收点波长进行测定,混合物溶液在这两个波长处得吸光度差值仅与咖啡因浓度成正比,而与苯甲酸钠浓度无关,可消除苯甲酸钠对咖啡因测定的干扰,从而实现咖啡因的定量分析: 咖啡因KC A =? 实验仪器与试剂 1.752Pro 型紫外可见分光光度计 2.标准咖啡因储备溶液(0.2500mg/ml ) 3.标准甲酸钠储备溶液(0.2500mg/ml ) 4.盐酸溶液(0.10mol/L ) 5.50ml 容量瓶12个 6.5ml 移液管4只 7.1cm 石英比色皿2个 8.安钠咖样品溶液 实验步骤 1.咖啡因标准系列溶液配制 按照下表配制咖啡因标准系列溶液 编 号 咖1 咖2 咖3 咖4 咖5 移取0.25mg/ml 标准咖啡因储备溶液体积 1.00ml 2.00ml 3.00ml 4.00ml 5.00ml 系列标准咖啡因溶液浓度 5μg/ml 10μg/ml 15μg/ml 20μg/ml 25μg/ml 说 明 以上溶液均用移液管移取至50ml 容量瓶中,用0.10mol/L 盐酸溶液稀释至刻度并摇匀。 2.苯甲酸钠标准系列溶液配制 按照下表配制苯甲酸钠标准系列溶液
4 紫外-可见分光光度法测定水中苯酚含量
实验二紫外-可见分光光度法测定水中苯酚含量 苯酚是工业废水中一种有害污染物质,需对水中酚含量控制。苯酚在270-295nm波长处有特征吸收峰,其吸光度与苯酚的含量成正比,应用Lambert-Beer定律可直接测定水中总酚的含量。 一、实验目的 1.学会使用Cary50型紫外-可见分光光度计 2.掌握紫外-可见分光光度计的定量分析方法 二、原理简介 紫外-可见吸收光谱是由分子外层电子能级跃迁产生,同时伴随着分子的振动能级和转动能级的跃迁,因此吸收光谱具有带宽。紫外-可见吸收光谱的定量分析采用朗伯-比尔定律,被测物质的紫外吸收的峰强与其浓度成正比,即: 其中A是吸光度,I、分别为透过样品后光的强度和测试光的强度,为摩尔吸光系数,b为样品厚度。 由于苯酚在酸、碱溶液中吸收波长不一致(见下式),实验选择在碱性中测试,选择测试的波长为288nm左右,取紫外-可见光谱仪波长扫描后的最大吸收波长。 Cary50是瓦里安公司的单光束紫外-可见分光光度计。仪器原理是光源发出光谱,经单色器分光,然后单色光通过样品池,达到检测器,把光信号转变成电信号,再经过信号放大、模/数转换,数据传输给计算机,由计算机软件处理。 三、仪器与溶液准备 1、Cary50型紫外-可见分光光度计 2、1cm石英比色皿一套 3、25 ml容量瓶5只,100 ml容量瓶1只,10ml移液管二支
配置250 mg/L苯酚的标准溶液:准确称取0.0250 g苯酚于250 mL烧杯中,加入去离子水20 mL使之溶解,加入0.1M NaOH 2mL,混合均匀,移入100 mL容量瓶,用去离子水稀释至刻度,摇匀。 取5只25 mL容量瓶,分别加入1.00、2.00、3.00、4.00、5.00 mL苯酚标准溶液,用去离子水稀释至刻度摇匀,作为标准溶液系列。 将溶剂,标准溶液,待测水样依此装入石英比色皿。按测试程序的提示,依次放入样品室中进行测试。 四、测试过程 1、确认样品室内无样品 2、开电脑进入Window 系统 3、点击进入Cary50 主菜单 4、双击Cary-WinUV图标 5、在Win-UV 主显示窗口下,双击所选图标“SCAN”以扫描测定吸收曲线:取上述标准系列任一溶液装进1cm石英比色皿至4/5,以装有蒸馏水的1cm石英比色皿作为空白参比,设定在220-350 nm波长范围内扫描,获得波长-吸收曲线,读取最大吸收的波长数据。 6、在Win-UV 主显示窗口下,双击图标“Concentration”进入定量分析主菜单 7、设定测试分析步骤: (l)单击Setup功能键,进入参数设置页面。在Wavelength处填入由步骤5获取的波长数据。 (2)按Cary Control 、Standards、Options、Samples、Reports、Auto store顺序,分别设置好菜单中每页的参数。按OK回到“Concentration”界面主菜单。 (3)单击View莱单,选择需要显示的内容。 例如基本选项Toolbar,buttons,Graphics,Report。 (4)单击Zero,提示“Load blank press OK to read” (放空白按OK读),放入空白蒸馏水到样品室内,按OK测试,测完取出样品。 (5)单击Start, 出现标准/样品选择页。选Selected for Analysis(选择分析的标准和样品)。此框的内容为准备分析的标准和样品。 (6)按OK进行分析测试。 依Presentstdl的提示:放入标准1然后按OK键进行读数。放标准2按OK进行读数。直到全部标准读完。 (7)出现“Present Samplel Press OK to read”提示框,根据提示,放入样品1按OK开始读样品,直到样品测完。 (8)可点击Save Method AS保存此方法,以后可以从Open Method调用此方法。从标准曲线读出水样中苯酚的含量(g/L),测试数据采用点击Save Data AS 保存。
分光光度法-生化实验
常用生化实验技术:分光光度法有色溶液对光线有选择性的吸收作用,不同物质由于其分子结构不同,对不同波长光线的吸收能力也不同,因此,每种物质都具有其特异的吸收光谱。有些无色溶液,虽对可见光无吸收作用,但所含物质可以吸收特定波长的紫外线或红外线。分光光度法主要是指利用物质特有的吸收光谱来鉴定物质性质及含量的技术,其理论依据是Lambert和Beer定律。 分光光度法是比色法的发展。比色法只限于在可见光区,分光光度法则可以扩展到紫外光区和红外光区。比色法用的单色光通过滤光片产生,谱带宽度为40~120nm,精度不高,而分光光度法则要求近于真正单色光,其光谱带宽最大不超过3~5nm,在紫外光区可到l nm以下。单色光通过棱镜或光栅产生,具有较高的精度。 一、光的基本知识 光是由光量子组成的,具有二重性,即不连续的微粒性和连续的波动性。波长和频率是光的波动性的特征,可用下式表示: λ=C/υ 式中λ为波长,具有相同的振动相位的相邻两点间的距离叫波长。υ为频率,即每秒钟振动次数。c为光速,等于299 770±4km/s。光属于电磁波。 自然界中存在各种不同波长的电磁波,列成表l-l所示的波谱图。分光光度法所使用的光谱范围在200nm~10μm (1μm=1 000nm)之间。其中200~400nm为紫外光区,400~760nm为可见光区,760~10 000 nm为红外光区。 二、朗伯一比尔(1ambert—Beer)定律 朗伯—比尔定律是比色分析的基本原理,这个定律是讨论有色溶液对单色光的吸收程度与溶液的浓度及液层厚度间的定量关系。此定律是由朗伯定律和比尔定律归纳而得。 1.朗伯定律一束单色光通过溶液后,由于溶液吸收了一部分光能,光的强度就要减弱:若溶液浓度不变,则溶液的厚度愈大(即光在溶液中所经过的途径愈长),光的强度减低也愈显著。 设光线通过溶液前的强度为Io(入射光的强度),通过液层厚为L溶液后.光的强度为I t(透过光的强度),则 表示透过光的强度是入射光强度的几分之几,称为透光度(transmittance),用T表示。透光度随溶液厚度的增
栀子双波长融合高效液相色谱数字化指纹图谱研究
栀子双波长融合高效液相色谱数字化指 纹图谱研究 (作者:___________单位: ___________邮编: ___________) 【摘要】目的采用双波长融合法建立栀子254 nm和326 nm波长下的融合HPLC指纹图谱。方法采用反相高效液相色谱法,使用CenturySIL C18 BDS柱(20 cm×4.6mm,5μm),以1%醋酸水-1%醋酸乙腈为流动相,低压线性梯度洗脱,柱温(30.0±0.15)℃,采用DAD检测器同时采集2个特征吸收波长下的信号,测定10批不同产地栀子HPLC指纹图谱。结果用“中药色谱指纹图谱超信息特征数字化评价系统”软件建立了栀子254 nm和326 nm的双波长融合HPLC指纹图谱,以栀子苷为参照物,确定了41个共有峰,并对其进行了全面评价。结论所建立的双波长融合谱能有效控制栀子的质量,克服了单波长检测时信息量有限的缺点,能较全面、真实地揭示中药材的内在质量特征。 【关键词】栀子;高效液相色谱数字化指纹图谱;双波长融合;超信息特征 Abstract:ObjectiveTo explore the HPLC-FPS of water extract
of Fructus Gardeniae by dual-wavelength fusing method. MethodsThe chromatographic fingerprints were obtained by injecting the sample solution each time on a CenturySIL C18 BDS column (20 cm×4.6 mm,5 μm) with the gradient elution solvent system composed of 1% acetate acid -water and 1% acetate acid -acetonitrile. The flow rate was 1.0ml/min, the column temperature was maintained at (30.00±0.15)℃and the DAD detector was used. ResultsThe HPLC-FPS of water extract of Fructus Gardeniae was established with dual-wavelength fusing method, acquired 41 common peaks by taking the peak of geniposide as referential peak. The fingerprints were comprehensively evaluated by “The digitized evaluation system of the Traditional Chinese Medicine fingerprint with the super-information characteristics”and the results were analyzed and compared with that of the fingerprints obtained under 265 nm. ConclusionThe dual-wavelength fusing method can effectively control the quality of Fructus Gardeniae and overcome the low information of mono-wavelength detection. It offers guarantee for the quality control of Fructus Gardeniae and provides a new method for thoroughly and authentically revealing the characteristics of TCM. Key words:Fructus Gardeniae; Dual-wavelength fusing method; High performance liquid chromatography;Fingerprints;
实验报告-紫外-可见分光光度法测铁的含量-
一、实验目的: 了解朗伯-比尔定律的应用,掌握邻二氮菲法测定铁的原理;了解分光光度计的构造;掌握分光光度计的正确使用方法;学会吸收曲线的绘制和样品的测定原理。 二、实验原理 邻菲啰啉是测定微量铁的较好试剂。在pH=2~9 的条件下,邻菲啰啉与Fe2+生成稳定的橙红色配合物,其反应式如下: Fe3+能与领二氮菲生成淡蓝色配合物(不稳定),故显色前加入还原剂:盐酸羟胺使其还原为Fe2+。。 三、仪器及试剂 紫外可见分光光度计、铁标准溶液:含铁0.01mg/mL、0.1%邻菲罗啉水溶液、10%盐酸羟胺水溶液、1mol/lNaAc缓冲溶液(pH4.6)。 四、实验步骤 1.吸收曲线的绘制和测量波长的选择 吸取0.0mL和6.0mL 铁标准溶液分别注入两个50 mL容量瓶中,依次加入5mlNaAc溶液,2.5ml盐酸羟胺溶液,5ml邻菲罗啉溶液,用蒸馏水稀释至刻度,摇匀。用1cm比色皿,以试剂空白为参比,在440~560nm之间,每隔0.5nm测吸光度。然后以波长为横坐标,吸光度A 为纵坐标,绘制吸收曲线,找出最大吸收波长。 2、标准曲线的绘制
分别吸取铁的标准溶液0.0、2.0、4.0、6.0、8.0、10.0ml于6只50ml容量瓶中,依次分别加入5ml醋酸-醋酸钠缓冲溶液,2.5ml盐酸羟胺溶液,5ml邻菲罗啉溶液,用蒸馏水稀释至刻度,摇匀,放置10分钟,在其最大吸收波长下,用1cm比色皿,以试剂溶液为空白,测定各溶液的吸光度,以铁含量(mg/50ml)为横坐标,溶液相应的吸光度为纵坐标,绘制标准曲线。 五、实验记录及数据处理 波长/nm 吸光度 标准溶液(0.01g/L)未知液容量瓶编号 1 2 3 4 5 6 7 吸取的体积0 2.0 4.0 6.0 8.0 10.0 吸光度A (1)绘制曲线图。
实验一-紫外分光光度法测定苯甲酸
实验一紫外分光光度法测定苯甲酸 一、实验目的 学习、了解紫外分光光度法原理 了解紫外分光光度计的结构和使用方法 二、实验原理 当辐射能(光)通过吸光物质时,物质的分子对辐射能选择性的吸收而得到的光谱称为分子吸收光谱。分子吸收光谱的产生与物质的分子结构、物质所在状态、溶剂和溶液的PH等因素有关。分子吸收光谱的强度与吸光物质的浓度有关。表示物质对光的吸收程度,通常采用“吸光度”这一概念来量度。 根据朗伯-比尔定律,在一定的条件下,吸光物质的吸光度A 与该物质的浓度C和液层厚度成正比。即A= LC 因此,只要选择一定的波长测定溶液的吸光度,即可求出该溶液浓度,这就是紫外-可见分光光度计的基本原理。 在碱性条件下,苯甲酸形成苯甲酸盐,对紫外光有选择性吸收,其吸收光谱的最大吸收波长为225nm。因此,采用紫外分光光度计测定苯甲酸在225nm处的吸收度就能进行定量分析。 三、仪器与主要试剂 TU-1810紫外可见分光光度计1cm石英比色皿 0.1M氢氧化钠溶液 苯甲酸(AR) 四、实验步骤 1、苯甲酸标准溶液的制备 称取苯甲酸(105℃烘干)100mg,用0.1M氢氧化钠溶液100ml溶解后,转入1000ml容量瓶中,用蒸馏水稀释至刻度.此溶液1ml含0.1mg 苯甲酸. 2、制作苯甲酸吸收曲线,选择最大吸收波长 ①移取苯甲酸标准溶液4.00ml于50ml容量瓶中,用0.01M氢氧化钠溶液定容,摇匀,此溶液1ml含苯甲酸8ug. 以氘灯为光源,用0.01M氢氧化钠溶液作为参比,改变测量波长(从210-240nm)测量8ug/ml苯甲酸的吸光度. ②以波长为横坐标,吸光度为纵坐标,绘制苯甲酸的紫外吸收曲线,并找出最大的吸收波长 (是否是225nm). 3﹑样品的测定 ①取10.00ml苯甲酸样品,放入50ml容量瓶中,用0.01M氢氧化钠
紫外分光光度计实验报告
UV-2550紫外分光光度计的使用和分光光度法测定对苯二酚姓名:XXX 专业:有机化学学号:312070303004 时间:2012.10.21 1.目的 (1)了解UV-2550紫外光谱仪的基本使用方法。 (2)了解测定对苯二酚的紫外光谱实验方法。 2. 试剂和仪器 2.1试剂: 标准溶液0.10m g/mL,准确称取0.25g对苯二酚溶于250ml容量瓶中,用水稀释至刻度,从中取出10ml于100ml容量瓶中,用水稀释至刻度,摇匀;pH=4.1的乙酸-乙酸钠缓冲溶液。 2.2 仪器: UV-2550型分光光度计。 3. 实验步骤 3.1 测量波长的选择 用吸量管吸取5.0ml对苯二酚标准溶液于25ml容量瓶中,加入0.5ml pH=4.1的乙酸-乙酸钠缓冲溶液,用二次蒸馏水定容,振荡混匀。15分钟后用1cm比色皿,275-330nm波长范围, 进行扫描。从吸收曲线上读出对苯二酚的最大吸收波长λmax。 3.2 对苯二酚含量的测定 (1)标准曲线的制作 在6个25ml容量瓶中,用吸量管分别加入0,1.0, 2.0, 3.0,4.0,5.0ml 对苯二酚标准溶液,加入0.5ml pH=4.1的乙酸-乙酸钠缓冲溶液,用二次蒸馏水定容,振荡混匀。用1cm比色皿,以试剂空白为参比溶液,在最大吸收波长处,用光度模块作标准曲线。 (2)试样中对苯二酚含量的测定 准确吸取一定体积的样品于40ml容量瓶中,加入0.5ml pH=4.1乙酸-乙酸钠,用水稀释至刻度,摇匀。在光度模块中直接读出试样中对苯二酚含量。 4. 实验结果 4.1 测量波长的选择 从吸收曲线上读出对苯二酚的最大吸收波长λmax=288.80。 见图1 吸收曲线 4.2 对苯二酚含量的测定 (1)标准曲线的制作 见图2 标准曲线 (2)试样中对苯二酚含量的测定 对苯二酚含量0.354 相对误差为11.5%
双波长分光光度法的基本原理及应用
双波长分光光度法的基本原理及应用 应用分光光度法对共存组分进行不分离定量测定时,通常采用的方法有双波长法,三波长法,导数光谱法、差谱分析法及多组分分析法等方法,其快速,简便的优点使这些方法在实用分析中得到越来越广泛的应用。其中以双波长法的应用为最多,该法的准确度和精密度要高于其它方法,是对共存组分不分离定量测定的有效方法之一。 实用中的双波长法主要采用等吸收波长法和系数倍增法两种分析方法,下面就其基本原理和应用作以介绍: 一、等吸收波长法 1、基本原理 图1是同一组分三个不同浓度供试液的吸收光谱图,经典分光光度法的定量测定通常是在被测组分的最大吸收波长处进行测定,根据兰伯一比耳定律,其吸光度值与被测组分的浓度C成正比,即: 依(3)式测定被测组分a,则可完全消除b组分的干扰,达到共存组分不分离进行定量测定的目的。 2、影响因素 (1)测定波长和组合波长的选择应使被测组分的△A值尽可能大,以增加测定的灵敏度和精确度。 (2)测定波长和组合波长应尽可能选择在光谱曲线斜率变化较小的波长处,以减小波长变化对测定结果的影响。 (3)干扰组分等吸收波长(组合波长)的选择必须精确,只有其△A值等于零时才能完全消除干扰,否则会引入测定误差。为此,在实用分析中,都是先配制一个干扰组分b的供试液,在仪器上准确找出等吸收波长,然后再对样品进行测定。 3 应用实例 等吸收波长法的一个典型应用实例为收载于《中华人民共和国药典》中的抗菌消炎药复方磺胺甲噁唑片的含量测定。复方磺胺甲噁唑片中含有磺胺甲噁唑(SMZ)和甲氧苄(TMP)两个成分,其吸收光谱见图3。 当测定SMZ时,选择其最大吸收波长257nm为测定波长,可以在干扰组分TMP的光谱曲线上304nm附近找到等吸收波长为组合波长消除其干扰;当测定TMP时,选择239nm为测定波长,可以在干扰组分SMZ的光谱曲线上295nm附近找到等吸收波长为组合波长消除其干扰,分别对SMZ和TMP进行含量测定。 二、系数倍增法 1 基本原理
双波长法测淀粉含量
附录4 直链淀粉和支链淀粉的测定(双波长法) 1、目的 淀粉一般都是直链淀粉和支链淀粉的混合物。直链淀粉和支链淀粉含量和比例因植物种类而不同,决定着谷物种子的出粉率和食物品质,并影响着谷物的贮藏加工。通过本实验学习掌握双波长测定谷物中直链淀粉和支链淀粉的含量。 2、原理 根据双波长比色原理,如果溶液中某溶质在两个波长下均有吸收,则两个波长的吸收差值与溶质浓度成正比。 直链淀粉与碘作用产生纯蓝色,支链淀粉与碘作用产生紫红色。如果用两种淀粉的标准溶液与碘反应,然后在同一个坐标系里进行扫描或做吸收曲线,即可达到实验目的。 3、仪器、试剂和材料 1、仪器 (1)电子分析天平 (2)分光光度计1台 (3)ph计 (4)容量瓶100mlx2,50mlx16 (5)吸管0.5mlx1,2mlx1,5mlx1 2、试剂 (1)乙醚 (2)无水乙醇 (3)0.5mol/LKOH溶液 (4)0.1mol/LHCL溶液 (5)碘试剂:称取碘化钾2.0g,溶于少量蒸馏水,在加碘0.2g,待溶解后用蒸馏水稀释定容至100ml。 (6)直链淀粉标准溶液:称取直链淀粉纯品0.1000g,放在100ml容量瓶中,加入0.5mol/LKOH10ml,在热水中待溶解后,取出加蒸馏水定容至100ml,即为1mg/ml直链淀粉标准溶液。 (7)支链淀粉标准溶液:用0.1000 g 支链淀粉按(6)法制备成1mg支链淀粉标准溶液。 3、材料 小麦粉 4、操作步骤 1、选择支链、直链淀粉测定的波长参比波长。 直链淀粉:取1mg/ml直链淀粉标准溶液1ml,放入50ml容量瓶中,加蒸
馏水30ml,以0.1mol/LHCL溶液调至PH3.5左右,加入碘试剂0.5ml,并以蒸馏水定容。静置20min,以蒸馏水为空白,用光束分光光度计进行可见光全波段扫描或用普通比色法绘出直链淀粉吸收曲线。 支链淀粉:取1mg/ml支链淀粉标准溶液1ml,放入50ml容量瓶中,加蒸馏水30ml,以0.1mol/LHCL溶液调至PH3.5左右,加入碘试剂0.5ml,并以蒸馏水定容。静置20min,以蒸馏水为空白,用光束分光光度计进行可见光全波段扫描或用普通比色法绘出支链淀粉吸收曲线。 2、制作双波长直链淀粉标准曲线:吸取1mg/ml直链淀粉标准溶液0. 3、0.5、0.7、0.9、1.1、1.3ml分别放入6只不同的50ml容量瓶中,加蒸馏水30ml,以0.1mol/LHCL溶液调至PH3.5左右,加入碘试剂0.5ml,并以蒸馏水定容。静置20min,以蒸馏水为空白,比色,吸光差值为纵坐标,直链淀粉含量(mg)为横坐标制备双波长直链淀粉标准曲线。 3、制作双波长支链淀粉标准曲线:吸取1mg/ml支链淀粉标准溶液2.0、2.5、3.0、3.5、4.0、4.5ml分别放入6只不同的50ml容量瓶中,加蒸馏水30ml,以0.1mol/LHCL溶液调至PH3.5左右,加入碘试剂0.5ml,并以蒸馏水定容。静置20min,以蒸馏水为空白,比色,吸光差值为纵坐标,支链淀粉含量(mg)为横坐标制备双波长支链淀粉标准曲线。 4、样品中直链淀粉、支链淀粉及总淀粉的测定:样品粉碎过60目筛,用乙醚脱脂,称取脱脂样品0.1g左右(精确到1ml),置于50ml容量瓶中。加0.5mol/LKOH溶液10ml,在沸水浴中加热10min,取出,以蒸馏水定容至50ml,静置。吸取样品液2.5ml两份(即样品液和空白液),均加蒸馏水30ml,以0.1mol/LHCL溶液调至PH3.5左右,样品中加入碘试剂0.5ml,空白液不加碘试剂,然后定容至50ml。静置20min,以样品空白液为对照比色。 五、结果处理 直链淀粉(%)=(X1*50*100)/(2.5*m*1000) 支链淀粉(%)=(X2*50*100)/(2.5*m*1000) 式中, X1----查双波长直链淀粉标准曲线得样品中直链淀粉含量(mg) X2----查双波长支链淀粉标准曲线得样品中支链淀粉含量(mg) m-----样品质量(g) 总淀粉(%)=直链淀粉(%)+支链淀粉(%)